CASE REPORT
Do not ignore multiple capillary malformations in a child
Malformações capilares múltiplas em criança
1Dermatovenereology Department of Coimbra University Hospital
2Genetic Department of Coimbra Pediatric Hospital, Coimbra, Portugal
Abstract
Capillary malformation-arteriovenous malformation syndrome (CM-AVM) is a rare autosomal dominant disorder caused by heterozygous mutations in RASA1 and EPHB4 that associates multifocal capillary malformations (CMs) with an increased risk for fast-flow vascular malformations (FFVMs) which can cause life-threatening complications.
We report the case of a 6-year-old girl with lip telangiectasia as well as multiple (>10) erythematous to brownish irregular macules and patches on the face, neck, trunk, and superior extremities from the age of 18 months with no local or systemic complaints. Several family members had similar lesions. Genetic testing revealed a heterozygous mutation in the EPHB4 gene, confirming the diagnosis of CM-AVM type 2. EPHB4 gene mutation was also present in her father and brother. No associated arteriovenous malformations (AVMs) were found after a thorough evaluation by cardiology and neurology, neither in the patient nor in the closest relatives.
The present case is a characteristic presentation of CM-AVM type 2 syndrome, with gradually appearance of multifocal CMs, lip telangiectasias, and a positive family history, without associated extracutaneous FFVMs. An increasing number of atypical CMs on the skin may be the only sign of CM-AVM and should alert the clinician to further investigate the presence of AVMs which can be life-threatening.
Keywords CM-AVM; Capillary malformation-arteriovenous malformation syndrome; Capillary malformations; Dermatology; RASA 1; EPHB4
Resumo
A Capillary malformation-arteriovenous malformation syndrome (CM-AVM) é uma doença autossómica dominante rara causada por mutações heterozigóticas nos genes RASA1 e EPHB4. Há malformações capilares (MCs) multifocais associadas a um risco aumentado de malformações vasculares de alto fluxo que podem causar complicações ameaçadoras de vida.
Uma menina de 6 anos apresentou-se com telangiectasias labiais e múltiplas (>10) máculas e manchas irregulares eritematosas-acastanhadas na face, pescoço, tronco e extremidades superiores, com início aos 18 meses de idade. Não havia queixas locais ou sistémicas associadas. O pai, irmão mais novo, a tia e avô paternos tinham lesões cutâneas semelhantes. O teste genético revelou uma mutação heterozigótica no gene EPHB4, confirmando o diagnóstico CM-AVM tipo 2. A doente foi avaliada por cardiologia e neurologia que excluíram malformações arteriovenosas (MAVs) associadas, e a doente mantem seguimento dermatológico e neurológico regulares, sem complicações até ao momento. O pai e o irmão foram também avaliados, tendo sido confirmada a mutação do gene EPHB4 e excluídas MAVs.
O presente caso é uma apresentação clássica de CM-AVM tipo 2, com aparecimento gradual de MCs multifocais, telangiectasias labiais e história familiar positiva, sem MAVs extracutâneas associadas. Com este caso pretendemos chamar a atenção que um número crescente de MCs cutâneas atípicas pode ser o único sinal de CM-AVM, que deve alertar o médico para a exclusão de MAVs potencialmente fatais.
Palavras-chave CM-AVM; Síndrome malformações capilares-malformações arteriovenosas; Malformações capilares; Dermatologia; RASA1; EPHB4
Introduction
Capillary malformation-arteriovenous malformation syndrome (CM-AVM) is a rare autosomal dominant disorder first described in 20031. It is caused by heterozygous mutations in RASA1 and EPHB4 genes, and associates multifocal capillary malformations (CMs) with an increased risk for fast-flow vascular malformations (FFVMs) (in the skin, muscle, bone, brain, and spine) that can be life-threatening2.
Case report
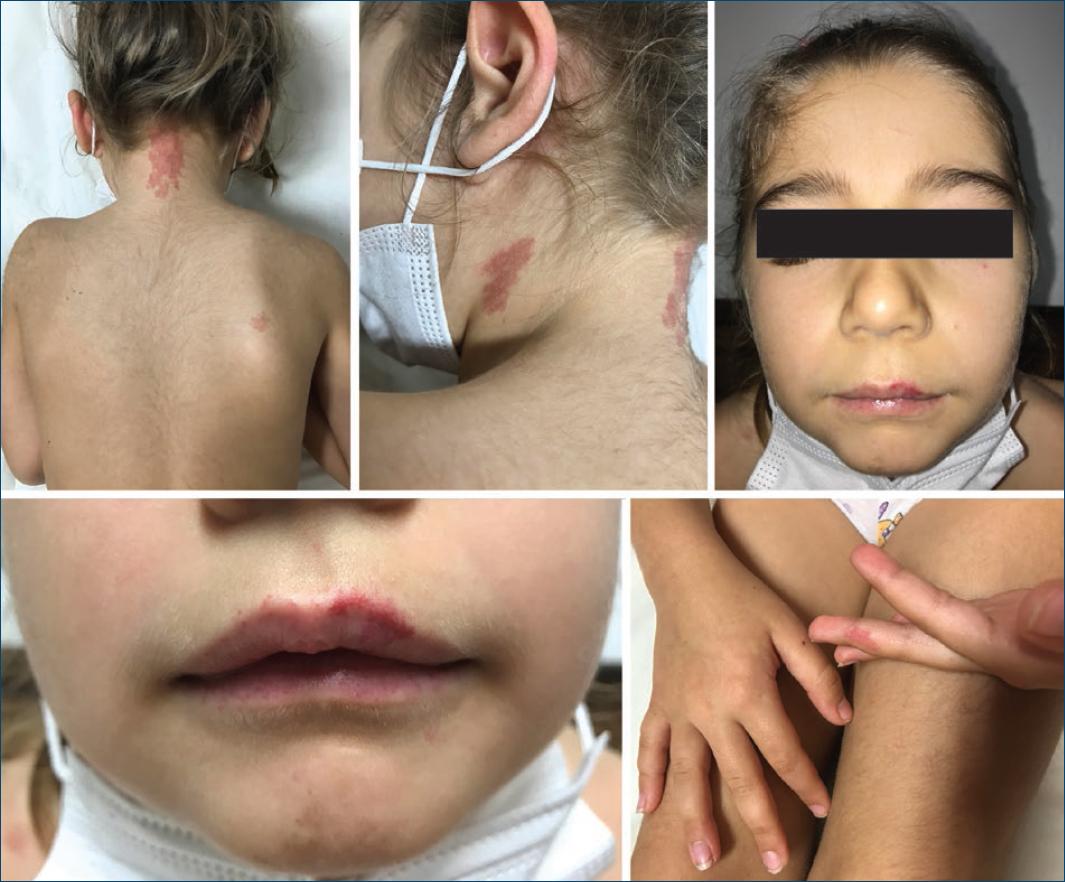
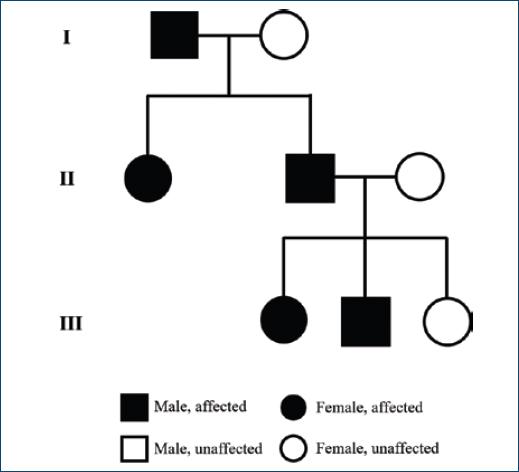
A 6-year-old girl presented with telangiectasias on the lips as well as multiple (>10) erythematous to brownish irregular macules and patches (largest 5 × 2 cm on the posterior neck), some surrounded by a whitish peripheral halo, located on the face, neck, trunk and superior extremities (Fig. 1). They had no thrill or increased temperature. Lesions started to appear at 18 months of age, progressively increasing in number over time. There were no local or systemic complaints. She was otherwise healthy. Her father had similar cutaneous lesions, as well as her younger brother, paternal aunt, and paternal grandfather (Fig. 2). Genetic testing revealed a heterozygous mutation in the EPHB4 gene, confirming the diagnosis of CM-AVM type 2. She was then referred to cardiology and neurology and performed directed studies (ECG, echocardiography, brain MRI) that did not reveal associated arteriovenous malformations (AVMs). New cutaneous lesions compatible with CMs continue to appear, and the patient maintains regular dermatology and neurology follow-up. No treatment is planned for her cutaneous CMs at this time. Her father and brother were also evaluated with confirmation of the mutation in EPHB4 gene and exclusion of AVMs.
Discussion
CM-AVM is a rare autosomal dominant inherited disorder that has been reported in association with heterozygous mutations in the RASA1 gene, which encodes the protein RASp21, and more recently to EPHB4, that signals via RAS/MAPK/ERK1/2 and PI3K/AKT/mTORC1 pathways3. Loss-of-function mutations in EPHB4 cause a similar vascular disorder to RASA1 mutations. The terms CM-AVM 1 and CM-AVM 2 have been proposed for mutations in RASA1 and EPHB4, respectively. CM-AVM 2 seems to be rarer than type 1. In a recent Spanish multicentre study including 64 patients with CM-AVM, only 4 out of 57 that were tested had EPHB4 mutations (and 73% were positive for RASA1 mutations)4.
When caused by mutations in EPHB4 gene, the phenotype, CM-AVM type 2, mimics RASA1-related CM-AVM type 1 but also hereditary haemorrhagic telangiectasia (HHT)3,5. However, CM-AVM patients do not have frequent epistaxis, the hallmark of HHT3.
Both type 1 and 2 CM-AVM are characterized by multifocal atypical CMs and increased risk for FFVMs. Cutaneous lesions are present in all patients and are generally multiple, pinkish macules or patches, that can exhibit a brownish hue, hypotrichosis, and a perilesional whitish halo. A “herald patch” (one macule/patch significantly larger than the others) has been recently described in 75% of the 64 patients studied by a Spanish group and can be a useful diagnostic clue4. CMs may be present at birth but usually gradually appear in early childhood6. FFVMs can present in the skin, muscle, bone, brain or spinal cord and rarely are multiple4.
Patients with CM-AVM 2 often show a pale central region in the largest skin macules, and a higher prevalence of Bier spots and telangiectasias (especially on the lips but also in the perioral region and on the upper thorax) that CM-AVM 1. Moreover, there is a lower association with FFVMs than in CM-AVM 1 (18% vs. 31%), of which only 3% are cerebral (compared to 10% in CM-AVM 1)3,4. Therefore, CM-AVM 2 seems to have a better outcome than type 14.
Evaluation of patients with multiple CMs is challenging because associated AVMs can be life threatening7. In addition to a complete family history and physical examination, other subspecialty evaluations may be necessary, especially neurology (to exclude potentially life-threating brain AVMs) and cardiology (since cardiac overload is a potential complication of FFVMs and may occur early in life)7. Valdivielso-Ramos et al. recently proposed brain and spinal MRI and genetic testing to all patients with ≥3 CMs, especially in familiar cases4.
The present case is a characteristic presentation of CM-AVM 2 syndrome, with progressive appearance of multifocal CMs, lip telangiectasias and a positive family history, without associated extracutaneous FFVMs. An increasing number of atypical skin CMs may be the only sign of CM-AVM and should alert the clinician to further investigate for potential life-threatening AVMs2,6. Moreover, close follow-up is warranted, even in patients with CM-AVM 24.
REFERENCES
1. Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73(6):1240–9. doi:10.1086/379793.
[ Links ]
2. Sibley CD, Ramien ML. Capillary malformation-arteriovenous malformation syndrome. JAMA Dermatol. 2019;155(6):733. doi:10.1001/jamadermatol.2019.0319.
[ Links ]
3. Amyere M, Revencu N, Helaers R, Pairet E, Baselga E, Cordisco M, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136(11):1037–48. doi:10.1161/CIRCULATIONAHA.116.026886.
[ Links ]
4. Valdivielso-Ramos M, Martin-Santiago A, Azaña JM, Hernández-Nuñez A, Vera A, Perez B, et al. Capillary malformation-arteriovenous malformation syndrome:a multicentre study. Clin Exp Dermatol. 2020:0-3. doi:10.1111/ced.14428.
[ Links ]
5. Yu J De, Streicher JL, Medne L, Krantz ID, Yan AC. EPHB4 mutation implicated in capillary malformation–arteriovenous malformation syndrome:A case report. Pediatr Dermatol. 2017;34(5):e227-30. doi:10.1111/pde.13208.
[ Links ]
6. CatalàA, RoéE, Vikkula M, Baselga E. Capillary malformation-arteriovenous malformation syndrome:a report of 2 cases, diagnostic criteria, and management. Actas Dermo-Sifiliográficas (English Ed.). 2013;104(8):710–3. doi:10.1016/j.adengl.2012.04.025.
[ Links ]
7. Weitz NA, Lauren CT, Behr GG, Wu JK, Kandel JK, Meyers PM, et al. Clinical spectrum of capillary malformation-arteriovenous malformation syndrome presenting to a pediatric dermatology practice:a retrospective study. Pediatr Dermatol. 2015;32(1):76–84. doi:10.1111/pde.12384.
[ Links ]
Copyright: © 2022 Portuguese Society of Dermatology and Venereology.












 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink