











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Alpha-1-antitrypsin is the major inhibitor of multiple serine proteases in the serum, which is mainly synthesized by hepatocytes and then secreted into the blood-stream to protect tissues from proteolytic damage. The protein is encoded by the SERPINA1 gene, located in the chromosome 14q32.1 region [1, 2].
Most pathogenic mutations in SERPINA1 result in the aggregation of misfolded proteins in the hepatocytes, causing liver injury through a toxic “gain of function.” On the other hand, the decrease in systemic AAT levels results in proteolytic lung damage by neutrophil elastase (“loss of function”) that predisposes to the development of early-onset panlobular emphysema and chronic obstructive pulmonary disease [1, 3].
The alpha-1 antitrypsin deficiency (AATD) is an auto-somal co-dominant condition and currently represents one of the most common and life-threatening genetic disorders. More than 150 variants of SERPINA1 have been reported, based on the migration speed of mutant AAT in an isoelectric field (e.g., medium-M, slow-S, and very slow-Z) [2, 4].
Pi*M constitutes the wild-type allele and is present in 85-90% of individuals [5]. Pi*Z (a substitution of lysine for glutamic acid at codon 342; rs28929474) and Pi*S (a substitution of valine for glutamic acid at codon 264; rs17580) are the most clinically relevant variants. Pi*Q0 alleles (null) represent a heterogeneous group of variants that yield no detectable protein in circulation.
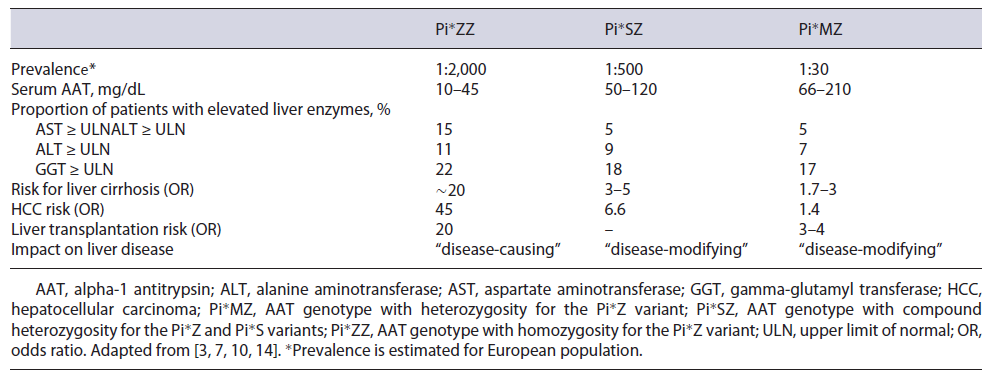
The classic severe AATD is caused in 90% of the cases by the homozygous Pi*Z variant (Pi*ZZ genotype) and affects approximately 1:2,000 individuals in Europe (Table 1). It is characterized by markedly decreased levels of serum AAT and confers a strong predisposition to lung and liver disease. Epidemiological studies show that the Pi*Z allele is more prevalent in Northern Europe, up to 8%, and Pi*S is more common in Southern Europe
(∼20%) [6]. The compound heterozygous genotype Pi*SZ affects 1:500 Caucasians, with intermediate AAT serum concentrations, and moderately increases susceptibility to lung disease and liver fibrosis [3].
In Portugal, the true prevalence of AATD remains unclear. However, it is estimated that 1:5,249 individuals have a Pi*ZZ genotype and 1:281 individuals have a Pi*SZ genotype [7, 8]. Meira L. et al., [8] based on a retrospective analysis, evaluated a cohort of Portuguese individuals tested for AATD between the years 2006 and 2015. The data accessible for the study comprised only AAT phenotyping or genotyping results, patient age at the time of diagnosis, and the health entities requesting the AATD genetic diagnosis. Unfortunately, clinical information or AAT serum levels were unknown. Overall, 1,684 individuals were considered, covering almost every region in Portugal. Most subjects were distributed into more common genotypes: Pi*MZ (25.4%), Pi*MS (15.5%), Pi*SZ (11.2%), Pi*ZZ (9.4%), and Pi*SS (5.6%). Different types of rare deficiency and null alleles were also detected.
Liver Disease in AATD
AATD-mediated liver disease is associated with the retention of misfolded AAT protein within the endoplasmic reticulum (ER) of hepatocytes that may result in liver fibrosis, cirrhosis, and hepatocellular carcinoma. Seventy percent of misfolded AAT protein is degraded by ER, 15% is secreted, and 15% forms insoluble polymers, which mainly persist within the endoplasmic reticulum as inclusions that are positive on periodic acid-Schiff staining and resistant to diastase (PAS-D) - the histologic hallmark of liver disease on biopsy [1, 9]. The AATD variants linked to polymerization, such as SIiyama, MDuarte, MMalton, and particularly the Z allele, may all show signs of liver injury [9].
Hepatopathy in AATD has a bimodal distribution, with the first peak in early childhood and the second peak typically after 50 years of age [10]. In the neonatal period, prolonged cholestasis is the most common clinical manifestation of liver injury. A Swedish neonatal screening program identified 120 babies with the PI*ZZ genotype in a population of 200,000 newborns. In this cohort, 12% had prolonged jaundice, and 8% of the neonates had severe liver disease [11]. Biochemical abnormalities were present in more than 50% of the Pi*ZZ neonates with spontaneous resolution within months, and most of them remained healthy at follow-up at age 18 years. Only about 3% had progressed to severe, life-threatening liver disease [12]. In the pediatric population, the AATD represents 3.5% of the causes of liver transplantation [13].
In adulthood, two large cross-sectional studies demonstrated the presence of significant liver fibrosis in 20%-36% of Pi*ZZ carriers, and the advanced liver fibrosis was 10-20 times more common in “Pi*ZZ” subjects compared with individuals without a “Pi*Z” mutation (non-carriers) [14, 15]. Although not fully identified, there is a strong influence of genetic and environmental modifiers in the development of liver disease in AATD. Male sex, age ≥50 years, obesity, metabolic syndrome, and diabetes mellitus were associated with liver fibrosis and primary liver cancer in this group of patients [10, 15]. A study involving 335 homozygous Pi*ZZ identified a single nucleotide polymorphism (SNP) that confers a higher risk for liver disease [16].
Notably, only 10% of patients with AATD develop cirrhosis, and 14.7% of adults who presented with AATD-related liver disease required transplantation. The overall incidence of hepatocarcinoma is 1.3%, and it seems similar to published data for aetiologies such as alcohol-related liver cirrhosis and primary biliary cholangitis [13]. A longitudinal study of Pi*ZZ individuals (n = 1,595) from the Swedish National AATD Register showed adjusted hazard ratios (HR) for hepatic and non-hepatic cancer of 23.4 and 1.3, respectively, in the Pi*ZZ individuals compared with the controls [17].
Regarding liver enzymes, as described in Table 1, only a small proportion of patients with AATD have abnormalities in liver parameters, which can fluctuate over time, even in patients with the Pi*ZZ genotype or with advanced liver fibrosis [18]. Schneider et al. [19] analyzed the liver phenotypes of a large cohort of patients with AATD (419 Pi*MZ; 309 Pi*ZZ; and 284 non-carriers). The authors concluded that gamma-glutamyl transferase (GGT) was the only parameter that clearly differed between the three genotypes, with the lowest values in non-carriers and the highest in Pi*ZZ subjects. This finding has been validated in another study [10]. Mean aspartate aminotransferase (AST) values were higher in Pi*ZZ versus Pi*MZ participants but were comparable between non-carriers and Pi*MZ individuals [19]. Accordingly, recent data from a large study showed that mean alanine aminotransferase (ALT) values were significantly higher in all AATD genotypes compared with non-carriers, and Pi*ZZ individuals had significantly higher AST values (adjusted odds ratio: 4.5) than any other assessed AATD subgroup [10].
Homozygous individuals for the Pi*S mutation (Pi*SS genotype) sheltered minimally elevated ALT values but no other hepatobiliary abnormality. However, Pi*SZ genotype displays higher liver enzymes and a clear predisposition to liver fibrosis/cirrhosis and primary liver cancer. This susceptibility is markedly lower than the one seen in Pi*ZZ carriers, which is consistent with the observed lower levels of intracellular polymers and a less pronounced lung disease [10, 19]. PI*MS genotype has not been consistently shown to increase the risk of liver disease [10].
On the other hand, the Pi*MZ genotype is primarily considered a disease-modifying factor in individuals with other liver diseases. Its relevance has been particularly well documented in individuals with cystic fibrosis as well as alcoholic/non-alcoholic fatty liver disease (ALD/NAFLD), in whom heterozygous Pi*Z presence greatly increased the odds to harbor cirrhosis. Pi*MZ was associated with a slightly elevation of liver enzymes and moderately increased odds for liver fibrosis/cirrhosis and cholelithiasis [3, 19]. Chen et al. [20] highlighted the role of Pi*MZ genotype as a risk factor for hepatic decompensation and the requirement for liver transplantation or liver-related death in a cohort of patients with compensated advanced chronic liver disease (576 patients with cirrhosis: 474 Pi*MM, 49 Pi*MZ, and 52 Pi*MS). Compared to the Pi*MM genotype, Pi*MZ was associated with increased rates of hepatic decompensation (hazard ratio 1.81) and liver transplant or liver-related death (hazard ratio 2.07). Moreover, a recent retrospective study identified the Pi*Z allele as an independent risk factor for liver transplantation and death in patients with advanced chronic liver disease (ACLD). In a population of 1,118 patients with ACLD, Pi*Z carriers (n = 42) had more severe portal hypertension and hepatic dysfunction, compared to non-carriers [21].
These data provide evidence about the profoundly detrimental impact of the Pi*Z allele on the outcomes of ACLD and ALD/NAFLD. As a significant proportion of Pi*MZ patients can have normal AAT levels, dosing serum levels are insufficient to identify routinely these patients. Genotyping significantly increases the costs for the routine assessment of ALD/NAFLD and ACLD patients. An approach with phenotyping would permit identifying these patients at a significantly lower cost. As Pi*MZ patients have an increased OR of up to 3 for liver cirrhosis, up to 1.4 for hepatocellular carcinoma, and up to 4 for liver transplantation (as referred in Table 1), routine phenotyping for A1ATD would permit tailoring the follow-up of this large group of patients according to their risk stratification at a reasonable cost. However, further multicenter studies to validate these findings are warranted.
Diagnosis of Liver Disease in AATD
AATD is a widely underdiagnosed condition. When clinically suspected, the diagnosis should be confirmed by the AAT serum level and the determination of the AAT phenotype and/or genotype. If the serum levels, phenotype, or genotype do not agree with each other or with clinical manifestations, then a rare or null variant should be considered [7, 9, 22]. AAT is an acute phase protein, which is why its serum levels may be falsely elevated during inflammatory and infectious processes.
The evaluation of liver disease is particularly important in patients with AAT variants that lead to polymerization of misfolded AAT protein in the ER (mostly Pi*ZZ carriers). Liver biopsy is not recommended as a method of diagnosis and follow-up, given its invasive nature, and should be employed in individuals with inconclusive non-invasive results and/or additional investigation. Non-invasive assessment based on blood tests and various elastography methods has been proposed to estimate liver fibrosis. Liver stiffness measurement (LSM) has proven to be useful in the diagnosis of liver fibrosis, especially to rule out advanced fibrosis [23]. Several studies in AATD-related liver disease showed promising results using this approach [14, 15, 18, 24-28].
Clark et al. [14] evaluated 94 non-cirrhotic Pi*ZZ adults and demonstrated that LSM by transient elastography (TE) represented the best parameter to identify advanced liver fibrosis (AUROC 0.92), with the cut-off of 8.45 Kpa having the highest accuracy. Moreover, GGT was the best one to detect significant fibrosis (F ≥2 on a 0-4 METAVIR scale) with an AUROC of 0.77, while TE, Fibrosis-4 (FIB-4), and AST-to-platelet ratio indices (APRI) were less well suited (AUROC 0.70/0.66/0.69, respectively).
Unfortunately, this cohort contained only 6 individuals with advanced liver fibrosis, and patients with cirrhosis were initially excluded [14]. The superiority of LSM by TE compared to blood tests in ruling out advanced liver fibrosis was also reported by Kümpers et al. [24].
Another large multinational study including 554 Pi*ZZ patients without known liver disease compared against 234 adults not carrying any AAT mutation confirmed the high prevalence of significant liver fibrosis in Pi*ZZ individuals (20%-36%). In this cohort, significant fibrosis was defined as LSM ≥7.1 kPa and advanced liver fibrosis defined as LSM ≥10.0 kPa. The last one was 9-20 times more frequent in Pi*ZZ individuals [15]. Also, the parallel assessment of TE, APRI, and HepaScore (age, sex, alpha2-macroglobulin, hyaluronic acid, bilirubin, and gamma-glutamyl transferase) values revealed a moderate correlation between the former two, whereas the correlations with the HepaScore were less robust.
Kim et al. [28] suggested that magnetic resonance elastography (MRE) may be accurate for identifying fibrosis (AUROC 0.9) in patients with alpha-1 antitrypsin deficiency. A MRE threshold of ≥3.0 kPa provided 88.9% accuracy, with 80% sensitivity and 100% specificity to detect the presence of any fibrosis (stage ≥1).
To note, the cut-offs used for detection of the corresponding fibrosis stages were different in several studies. The cut-off of LSM ≥5.45 kPa for significant liver fibrosis suggested by Clark et al. [14] does not seem to be useful for risk stratification as these values are seen in >50% of Pi*ZZ patients [14, 24]. Since the etiology of liver disease has an important impact on LSM, further studies are needed to validate the best LSM cut-off for screening of liver disease in AATD.
The recently published studies revealed other interesting features of Pi*ZZ-related liver disease. Controlled attenuation parameter (CAP) ≥280 dB/m, suggesting severe steatosis (grade 3), was detected in 39% of Pi*ZZ carriers versus 31% of controls, and about 44% of Pi*ZZ carriers displayed histological liver steatosis [14, 15]. Alterations in lipid metabolism observed in Pi*ZZ individuals (e.g., lower serum levels of triglycerides, very low-density lipoproteins, and low-density lipoproteins compared with controls) indicate that impaired hepatic secretion of lipids might play a role. However, it is important to mention that the accuracy of CAP for predicting histological steatosis has not been validated in AATD. The authors suggest in Figure 1 a diagnosis approach flowchart for patients with suspicion AATD-liver disease.
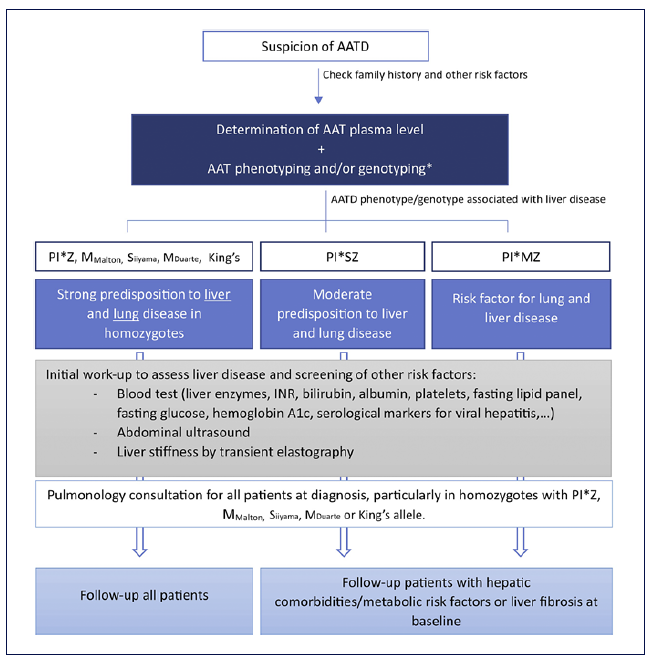
Fig. 1 The authors suggest the following diagnostic approach flowchart to assess liver disease in patients with AATD (*or whole gene sequencing depending on availability and/or the need for more detailed interpretation). The surveillance of Pi*MZ and Pi*SZ subjects needs to be adjusted to the overall clinical context that includes the presence of hepatic comorbidities/metabolic risk factors, other genetic factors as well as the presence/absence of baseline liver fibrosis as evaluated by non-invasive methods. AAT, alpha-1 antitrypsin; AATD, alpha-1 antitrypsin deficiency; PI*MZ, AAT genotype with heterozygosity for the Pi*Z variant; Pi*SZ, AAT genotype with compound heterozygosity for the Pi*Z and Pi*S variants.
Treatment of Liver Disease in AAT
For the treatment of AATD-related lung disease, the Food and Drug Association approved in 1987 the intra-venous augmentation therapy with plasma-purified AAT, the first disease-specific therapy for AATD. In the last years, several trials showed a significant reduction in the annual rate of lung density and, thereby, a decrease in the progression of lung emphysema while on AAT augmentation [7, 22, 29, 30].
Unfortunately, for endstage liver disease related to AATD, the liver transplantation is the only available curative treatment, with good survival and long-term outcomes for both children and adults [13, 31, 32]. Until the time of writing, there is no robust evidence of non-pharmacological interventions, such as smoking cessation or weight loss on liver outcomes. According to the known pathophysiology, it is expected that both of these behaviors would improve liver outcomes. Regarding this subject, previous data suggest that breastfeeding may have a protective effect in severe liver disease and early death in infants with alpha 1-antitrypsin deficiency [33].
Future Directions
The knowledge of pathological mechanisms of liver disease in AATD led to the investigation of several approaches for treating AATD-related liver disease. Some of these strategies are currently under evaluation in early-phase clinical trials and others in preclinical stages [1, 3, 34, 35]. Therapeutic targets address different steps of production, secretion, and elimination of misfolded AAT protein in the hepatocytes.
Silencing the production of mutated AAT constitutes a promising approach in the treatment of AATD-related liver disease. The small-interfering RNAs (siRNAs) andthe antisense oligonucleotides (ASOs) are the most common strategies in this field that modulate gene expression by inducing enzymatic degradation of targeted mRNA and consequently interrupting the production of their corresponding proteins [34].
Turner et al. [36] showed the first evidence of siRNA therapeutic designed to silence expression of Z-AAT (the Z variant of AAT) mRNA (Table 2). His study demonstrated a deep and durable knockdown of hepatic AAT production based on an observed reduction in serum AAT concentrations with no occurrence of major adverse events or hepatotoxicity in patients with PI*ZZ and healthy volunteers. Despite the results, the study was terminated early because of toxicity findings related to the delivery vehicle (ARC-EX1) seen in a non-human primate study.
Table 2 Overview of the most important clinical studies listed in ClinicalTrial.gov for therapy of liver disease in AATD
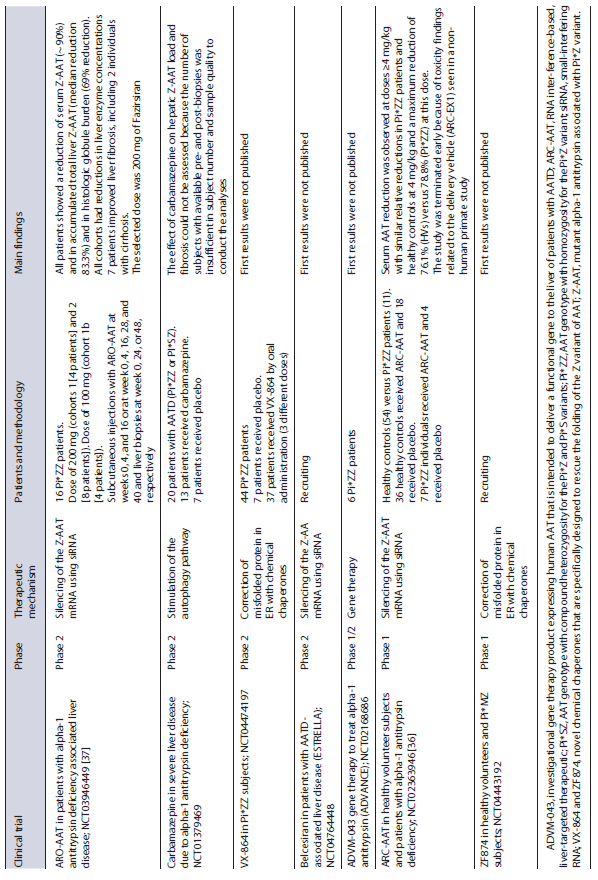
Recently, the ARO-AAT2002 open label trial (NCT03946449) evaluated the safety and efficacy of ARO-AAT injection (hepatocyte-targeted siRNA therapeutic against Z-AAT mRNA, Fazirsiran) administered subcutaneously to patients with alpha-1 antitrypsin deficiency. The Pi*ZZ patients received subcutaneous injections with ARO-AAT and underwent liver biopsies at weeks 0 and 24 or 48. The first results showed a reduced serum and liver Z-AAT and histologic globule burden in all patients and decreased serum ALT and GGT. Moreover, 7 individuals displayed an improvement in liver fibrosis, including 2 individuals with cirrhosis at baseline. These data demonstrate that removal of the causative factor, Z-AAT, in AATD liver disease ameliorates liver injury and can lead to an improvement in fibrosis [37]. This trial was followed by double-blindphase 2 clinical trial which results will be published soon. Phase 3 with this molecule (Fazirsiran) will begin recruiting in the end of 2022. Other trials based on siRNAs therapeutic that are currently ongoing are shown in Table 2. This approach seems to be a logical choice for individuals with isolated, advanced liver disease, but the long-term impact of the resulting decrease in serum AAT levels remains an important concern.
The administration of ASO can also represent a possible future direction in the treatment of liver disease [38, 39]. Some data from studies in preclinical stages showed that administration of ASO in Pi*ZZ transgenic mice led to an approximately 80% reduction in levels of circulating normal AAT and ameliorates liver fibrosis [38].
The correction of misfolded proteins in the ER with chemical chaperones is one of the approaches that have been studied. Burrows et al. [40] demonstrated in experimental models that phenylbutyrate (PBA), a substance used for treatment of urea cycle disorders, increased five-fold the secretion of functionally mutant AAT in Pi*ZZ mice. However, in a preliminary study of a small number of patients with AATD (9 Pi*ZZ individuals and 1 with a homozygous null genotype), the oral administration of PBA during 14 days was not effective in increasing AAT blood levels, and metabolic side effects were noted [41]. As shown in Table 2, other folding correctors are the object of study in current trials, such as VX-864 and ZF874.
Some studies have identified small molecules that are effective in reducing the polymerization of Z-AAT in vitro [34, 35, 42]. Recently, Lomas et al. [43] described a small molecule termed “GSK716” that blocked intracellular polymerization of Z-AAT and increased the circulating levels of monomeric protein by sevenfold in a transgenic mouse model of AATD.
On the other way, intracellularly expressed antibody fragments (“intrabodies”), particularly ones that consist of one heavy and one light variable domain linked by a synthetic flexible peptide, were also used to block the polymerization in the ER. scFv is capable of preserving antigenbinding specificity and can be targeted to subcellular compartments by incorporating trafficking signals specific for the ER [34, 35]. In vitro, the scFv4B12 intrabody reduced the intracellular polymerization of Z-AAT up to 60% and increased its secretion, keeping the functional activity of AAT against neutrophil elastase [44].
Another pathway in decreasing the hepatic burden of AAT is autophagy, which is able to degrade structures that are too large to be processed via the proteosome, namely the misfolded proteins. Drugs approved and used for other diseases, such as carbamazepine and rapamycin, have been found to be promoters of autophagy and thereby reduce the number of intra-hepatic Z-AAT inclusions and hepatic fibrosis in a mouse model of AATD-associated liver disease [45, 46]. A recent randomized, double-blind, and placebo-controlled trial (NCT01379469) evaluated the effect on hepatic Z-AAT load and liver fibrosis of a 52-week treatment with carbamazepine in individuals with Pi*MZ/Pi*ZZ and severe liver disease. However, the main outcomes could not be assessed because the number of subjects with available pre- and post-biopsies was insufficient in subject number and sample quality to conduct the analyses (Table 2).
The advance of stem cells and molecular biology techniques allowed the development of promising therapeutics for liver disease in AATD. The recent study of Baligar et al. [47] showed that the transplantation of human mesenchymal stem cells and bone marrow-derived stem cells in transgenic mice expressing human Z-AAT confer some competitive advantages compared to host cells that could lead to pathological improvement. Transplantation of these cells resulted in the decline of globule-containing hepatocytes and partially improved liver pathology as reflected by inflammatory response, fibrosis, and apoptotic death of hepatocytes. Previously, Yusa et al. [48] accomplished the biallelic correction of the underlying mutation causing the expression of Z-AAT in the genome of human induced pluripotent stem cells derived from Pi*ZZ patients via a combination of piggyBac technology and zinc finger nucleases. This gene correction was sustained in subsequently differentiated hepatocyte-like cells and led to a restored structure and enzymatic function of native AAT.
Viral vector-mediated expression of short hairpin RNAs can be efficiently used to knockdown and functionally evaluate disease-related genes in patient-specific pluripotent stem cells. These methodologies can achieve a relevant reduction (−66%) of intracellular Pi*Z protein in hepatic cells after differentiation of patient-specific pluripotent stem cells [49].
Conclusion
Liver involvement constitutes the second most common clinical manifestation and cause of death in patients with AATD [3]. The assessment of liver fibrosis is a crucial step in the evaluation and follow-up of these patients. Among the non-invasive methods, the LSM by TE has particular interest and is useful to identify advanced liver fibrosis. Currently, liver transplantation is the only curative therapy for AATD-related liver disease. However, several promising strategies are currently under investigation and may emerge in the near future. In the light of clinical studies, silencing the production of mutated AAT using small-interfering RNAs seems to take advantage when compared with others strategies.

