











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Colorectal cancer (CRC) is the second most common cause of death from cancer worldwide [1]. Despite advances in screening and treatment, survival at 5 years remains only 65% [2]. Understanding the specific mechanisms of tumorigenesis may be critical for an optimal management of this disease, via prognostication and identification of new and more specific targets with the hope of defining individualized therapies.
Three main carcinogenic pathways of CRC have been recognized through the identification of major genetic events. The chromosomal instability pathway is characterized by tumours harbouring a RAS mutation, the mutator pathway can be identified via MLH1, MHS2, PMS2 and/or MSH6 silencing and the methylator pathway, by the fact that most tumours harbour a BRAF mutation and MLH1 is silenced by gene promoter hypermethylation [3].
The presence of activating mutations in RAS has been associated with a poorer prognosis [4] and is recognized as a strong predictor of resistance to EGFR-targeted agents [5, 6]. For these reasons mutational analysis of the RAS proto-oncogene is routinely performed in metastatic CRC in order to predict their response to targeted therapies - cetuximab or panitumumab.
The presence of microsatellite instability (MSI) identifies tumours from the mutator pathway with specific phenotypic features such as tumour location in the right colon, mucinous differentiation, “Crohn-like” lymphocyte responses or tumour-infiltrating lymphocytes, and better prognosis [7, 8]. Furthermore, stage II microsatellite instability-high (MSI-H) tumours with high-risk features for local or distant recurrence may not benefit from adjuvant chemotherapy due to resistance to fluoropyrimidines [9, 10]. Tumours with MSI exhibit some overlapping features with those emerging from the methylator pathway, and approximately 40% of tumours with an MSI phenotype may be associated with the so-called “serrated” pathway of CRC, where MLH1 silencing occurs through hypermethylation. This model of progression to cancer is somewhat accelerated as compared to the suppressor pathway involved in the classic adenoma-carcinoma sequence. From a molecular point of view, there is a higher incidence of BRAF mutations, whereas TP53 mutations display a lower rate [11]. BRAF mutations have been identified in metastatic CRCs with wild-type, non-mutated KRAS. These tumours seem to have a worse prognosis and may be resistant to anti-EGFR therapies [12].
The identification of these different pathways has brought about the recognition that CRC is no longer a homogeneous disease - different phenotypes having different behaviours, prognoses and responses to therapy [13].
Against this background and using a large, retrospective, single-institution series of cases with at least 4 years of follow-up, we evaluated clinical, morphological and genetic characteristics of CRC to assess their impact on clinical outcome, in a real-life scenario.
Methods
Patient Population
This retrospective cohort study included 242 consecutive patients with CRC diagnosed and treated between 2012 and 2014 (Hospital Beatriz Angelo). Only cases with available surgical specimens of the primary tumour were included. We excluded patients (i) with unresectable M1 disease or who underwent urgent surgery due to colonic obstruction and (ii) patients undergoing endoscopic resection. An electronic database was created in REDCap. Demographic, clinical and therapeutic data were collected from medical records. Patients were staged according to the American Joint Committee on Cancer/Union for International Cancer Control (AJCC/UICC) tumour node metastasis (TNM) staging system (7th edition). The study was approved by the hospital Ethics Committee.
Immunohistochemical and Molecular Analysis
Surgical specimens were retrieved and centrally reviewed. CRCs were analysed for RAS status, BRAF inactivation and immune expression of MMR genes.
Both RAS and BRAF status were assessed by real-time polymerase chain reaction (real-time PCR) in formalin-fixed, paraffin-embedded tissue sections. For KRAS and NRAS we analysed exons 2, 3 and 4. For BRAF, exon 15 was analysed. Only RAS wild-type tumours were tested for BRAF mutations.
MMR deficiency (dMMR) was characterized by the absence of MLH1, MSH6, MSH2 and/or PMS2 expression, evaluated in tissue microarrays. Staining of MMR proteins was performed using a Ventana Benchmark Ultra automated staining system, according to the manufacturer instructions, using the following monoclonal antibodies: anti-MLH1 (M1, MM PAB-US), anti-MSH2 (G219-1129, MM PAB-US), anti-MSH6 (SP93, RM PAB-US) and anti-PMS2 (A16-4, MM PAB-US). Normal colonic crypt epithelium adjacent to the tumour and stromal cells served as internal positive controls. Loss of expression was recorded when nuclear staining was absent in malignant cells but preserved in normal epithelial and stromal cells. All the tissue microarrays were independently evaluated by 2 pathologists. The few cases with discrepant scoring were re-evaluated by a third pathologist. Tumours with MLH1 or PMS2 methylation and concomitant BRAF mutation were considered to be part of the methylator pathway. In our hospital, routine evaluation of MMR gene expression by immunohistochemistry only started in 2016 and, thus, dMMR tumours (without BRAF mutations) were only identified when this study was performed.
Tumour genotyping was attempted in all cases but, for technical reasons, MMR status was informative in only 198/242 cases, RAS in 241/242 and BRAF in all cases tested (134/134 RAS wild type).
Tumour Location
Primary tumours originating distally from the splenic flexure were classified as left-sided tumours (sigmoid and descendent colon, including splenic flexure), whereas those originating proximally were right-sided (appendix, caecum, ascending colon, hepatic flexure and transverse). Rectal tumours were analysed separately.
Study Outcomes
Primary clinical outcomes were evaluated in terms of overall survival (OS) and disease-free survival (DFS). OS was defined as the time from surgery until death or last follow-up. DFS was the time from surgery to the first radiographic or endoscopic evidence of relapse (new colonic tumour, including both anastomosis relapse or metachronous CRC tumours, or metastasis). Survival data were censored at the time of the last follow-up.
Statistical Analysis
Categorical variables were compared using χ2 analysis. Continuous variables were compared by a two-tailed Student t test or Mann-Whitney U test according to normal and non-normal distributions. Kaplan-Meier survival curves were plotted to assess the temporal rate of events (OS, DFS). The log rank test was used to compare survival-free durations. Multivariable models (Cox regression) were adjusted for stage at diagnosis, primary tumour location, age and molecular profile status (MMR, BRAF and RAS). We also performed multivariable analysis including adjuvant chemotherapy in stage III CRC. All statistics were performed using SPPS (version 23).
Results
Overall, 242 patients were included with curative intent colorectal surgery at Hospital Beatriz Ângelo, between 2012 and 2014. There was a slight male predominance (57.4%), and the mean age at diagnosis was 69.3 ± 12.9 years. The median follow-up was 49 months (interquartile range 34-58; OS 4 years, DFS 3.5 years). Disease stages were the following: I 20.7%, II 32.6%, III 33.5% and IV 13.2%. In upfront stage IV, most patients had liver metastasis (68%), followed by the peritoneum (25%) and lung (7%). During follow-up, 34.7% of patients died (patients without metastasis 29%; patients with metastasis 72%). The rate of relapse in upfront stage I-III patients was 18.6%, compared to 44% in stage IV. 50% of CRC patients without metastases received adjuvant chemotherapy (single agent capecitabine 35.3% and fluoropyrimidine-oxaliplatin combination 64.7%). Age and comorbidities influenced the choice of the chemotherapy regimen (capecitabine alone vs. doublet schemes). Nevertheless, age by itself was not a limiting factor to prescribe adjuvant chemotherapy. In patients with metastases, 84.4% were treated with chemotherapy, namely 5-fluorouracil/leucovorin (5-FU/LV) plus oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) with bevacizumab or cetuximab. In rectal cancer, neoadjuvant chemotherapy regimens included doublet regimens (capecitabine plus oxaliplatin or 5-FU/LV plus oxaliplatin) and were prescribed in 43.2% of patients.
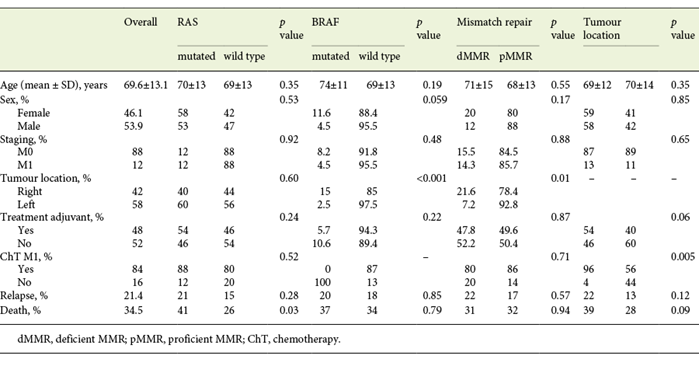
Patient and tumour characteristics are summarized in Table 1. The proportion of RAS-mutated tumours was 55.6%. BRAF inactivation occurred in 7% of all cases, and 15.6% had dMMR.
Table 1: Colonic tumours: clinical and demographic data according to tumour location, RAS, BRAF and MMR status
Colonic Tumours
Colonic and rectal tumours were analysed separately as they are considered 2 different entities with different prognosis and treatment protocols. Although we had 206 cases of colonic tumour, genotyping was possible in 170/206 cases only.
Overall, 54.8% (113/206) of colonic tumours were RAS mutated and were not evaluated for BRAF mutations. 15.8% (27/170) were dMMR and 7.8% were BRAF mutated (16/206). Concerning dMMR tumours, 22% (6/27) were BRAF mutated and considered to belong to the methylator pathway. The remaining 21 were offered germline mutation analysis to identify hereditary cases, and so far no germline mutations have been identified.
Clinical Features
Table 2 summarizes univariate analysis for clinical and molecular biomarkers. Age and TNM stage were the most discriminant variables both for DFS and OS. In respect to tumour location, most tumours were left-sided (58%). No differences were found in age (69 vs. 70, p = 0.35), gender distribution (male 58 vs. 42%, p = 0.85), synchronous metastases (13 vs. 11%, p = 0.65), relapse rate (22 vs. 13%, p = 0.12) or mortality rate (39 vs. 28%, p = 0.09) between left- and right-sided tumours. In stage I-III CRC, there was a trend towards a better prognosis in right-sided tumours both for DFS and OS (Fig. 1a, b).
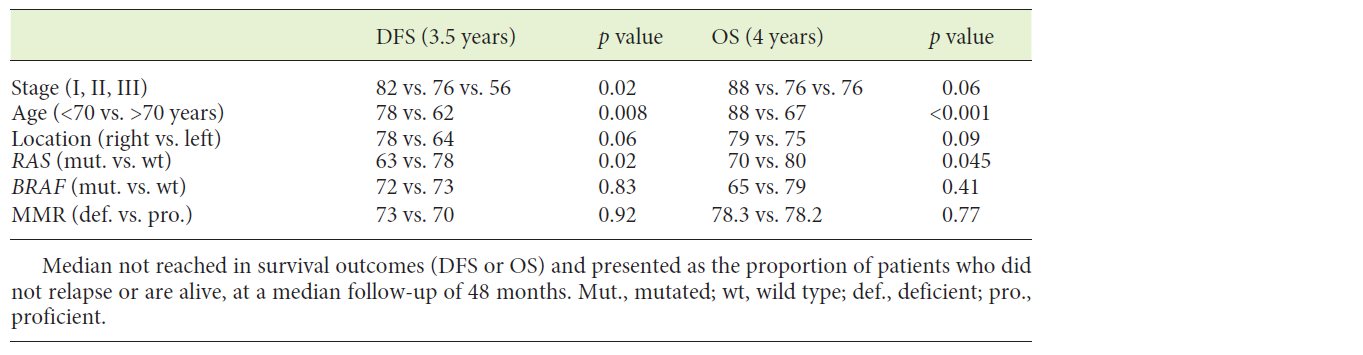

Fig.1: Kaplan-Meyer plots comparing right- and left-sided tumours in patients without metastasis. a Overall survival in CRC patients without metastasis (median not reached; at a median time point of 48 months: 79 vs. 75%, p = 0.09). b Disease-free survival (median not reached; at a median time point of 48 months: 78 vs. 64%, p = 0.06).
Molecular Profile
BRAF-mutated tumours (16/206) were more frequent in the right colon (15 vs. 2.5%, p < 0.001), with a trend towards a higher prevalence in female patients (11.6 vs. 4.5%, p = 0.059). dMMR tumours were also predominantly right-sided (21.6 vs. 7.2%, p = 0.01) whereas no differences were found in the distribution of RAS-mutated tumours (Table 1).
In stage I-III patients, RAS-mutated tumours had a worse median DFS (p = 0.02) and a lower median OS compared to RAS wild-type ones (p = 0.045; Table 2, Fig. 2a, b). BRAF mutation and dMMR did not influence DFS or OS (Table 2). After relapse, 21/33 patients died during the follow-up. 3/21 had BRAF-mutated tumours. Time to death after relapse was 3.5 months in cases of BRAF-mutated tumours and 18.6 months in BRAF wild-type ones (p = 0.17). Although the low number of BRAF-mutated relapsed patients precludes a survival analysis, the length of survival is clearly lower in patients with BRAF-mutated tumours.
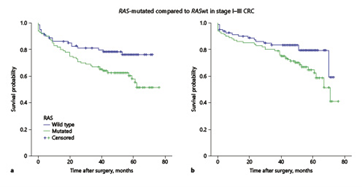
Fig.2: Kaplan-Meyer plots comparing RAS-mutated and RAS wild-type (wt) tumours in patients without metastasis. a Overall survival in CRC patients (median not reached; at a median time point of 48 months, 70 vs. 80%, p = 0.045). b Disease-free survival (median not reached; at a median time point of 48 months: 63 vs. 78%, p = 0.02)
In patients with synchronous metastases, no difference in median OS was found according to RAS status (16 vs. 46 months, p = 0.53) or dMMR (11 vs. 32 months, p = 0.29); only 1 patient had a BRAF-mutated tumour which precluded any statistical analysis.
Multivarable Analysis
Table 3 shows multivariable analysis for DFS and OS in patients with non-metastatic disease. In a model including age, stage at diagnosis and molecular profile (RAS, BRAF and dMMR), we observed that increased age and disease stage were associated with lower DFS. Patients with RAS-mutated tumours also had a marginally lower DFS (aHR 1.77, 95% CI 0.96-3.27, p = 0.07). Similarly, OS was also significantly associated with increased age and disease stage, whereas patients with RAS-mutated tumours also had a trend towards lower survival (aHR 1.91, 95% CI 0.96-3.77, p = 0.07). In stage III CRC, adjuvant chemotherapy showed a trend to a higher OS (aHR 0.45, 95% CI 0.16-1.11, p = 0.08). Molecular biomarkers did not influence survival outcomes in this subset of patients, most probably due to the low number of patients (online suppl. Table 1; for all online suppl. material, see www.karger.com/doi/10.1159/000516009).
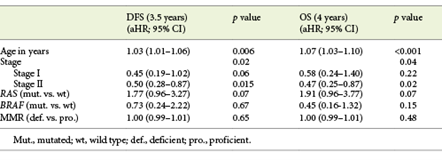
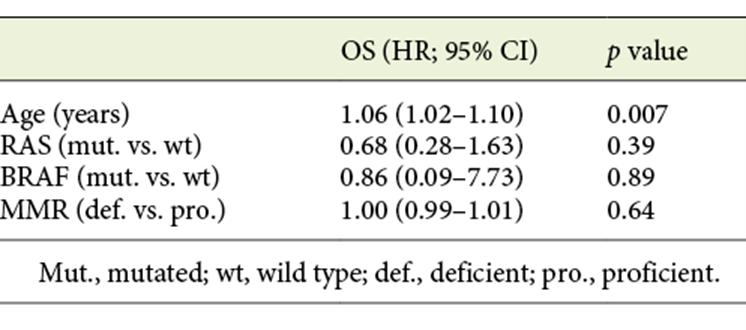
Table 4 shows multivariable analysis for patients with metastases. Age was the only factor associated with worse OS (HR 1.06, 95% CI 1.02-1.10, p = 0.007). As previously reported, at least 4 different chemotherapy regimens were prescribed in patients with metastasis. Moreover, patients changed to 2nd and 3rd lines of chemotherapy. Therefore, chemotherapy regimens were not included in the multivariable analysis in this subgroup of patients.
Rectal Tumours
A total of 36 rectal tumours were included, of which 8 had synchronous metastases at diagnosis. Excluding metastatic patients, 21.4% (6/28) relapsed and 28.6% (8/28) died during follow-up. The RAS mutation was present in 60.7% (17/28), BRAF in 3.6% (1/28) and dMMR in 10.7% (3/28). Of the 6 patients who relapsed, 4 had a RAS mutation and none had dMMR. Concerning the 8 patients who died, 5 had a RAS mutation and 1 had dMMR. Response to chemoradiotherapy was not significantly associated with RAS mutation (60 vs. 75%, p = 0.54) or dMMR (50 vs. 67%, p = 0.60)
Discussion
We evaluated the frequency and prognostic value of RAS and BRAF mutations and dMMR in a retrospective real-life cohort of consecutive patients with CRC who underwent surgery for the primary tumour in a single institution, including both metastatic and non-metastatic cases with at least 4 years of follow-up.
In patients without metastases, the RAS mutation had a negative impact on both DFS (p = 0.02) and OS (p = 0.045). After adjusting for age, stage at diagnosis and molecular biomarkers, RAS-mutated tumours still had a trend towards worse survival outcome, due to a higher probability of relapse (DFS: aHR 1.77, 95% CI 0.96-3.27, p = 0.07) and death (OS: aHR 1.91, 95% CI 0.96-3.77, p = 0.07). Prior studies in stage III colon cancer showed that KRAS exon 2 mutations are associated with worse outcome, largely due to early recurrence [14, 15]. In the RASCAL II study, mutation in KRAS was associated with a worse overall survival (HR 1.23, 95% CI 1.08-1.54, p = 0.008) and failure-free survival (HR 1.3, 95% CI 1.09-1.54, p = 0.004).
In the present study, the prevalence for BRAF inactivation was low (7.8%) compared to previous published data (8-14%) [13]. BRAF-mutated tumours were more frequent in the right colon, but no differences in OS or DFS were found. Conflicting results have been published on the prognostic value of BRAF mutation in stage I-III CRC [13]. In a multicentre study, including 2,246 patients with stage II-III colon cancer, BRAF mutation was not a prognostic factor (HR 0.93, 95% CI 0.73-1.20, p = 0.58) [16]. Nevertheless, Gavin et al. [17] reported lower OS and survival after relapse in BRAF-mutated tumours, but no difference in DFS in early-stage disease. In our series, we also found that, after relapse, BRAF-mutated tumours seem to have a lower survival (median 3.5 months) compared to BRAF wild-type ones (median 18.6 months) (NS probably due to low numbers). Furthermore, BRAF mutational status may influence the response to chemotherapy after relapse. In former trials [18, 19], a triplet chemotherapy regimen (5-FU, irinotecan and oxaliplatin - FOLFOXIRI), with or without bevacizumab, resulted in a survival benefit over a doublet regimen of 5-FU and irinotecan (FOLFIRI), with or without bevacizumab, in BRAF-mutated metastatic CRC. In clinical practice, intensifying first-line chemotherapy in fit patients with synchronous or metachronous metastatic BRAF-mutated CRC is now preferentially recommended. In the present series only 3 patients with BRAF-mutated tumours relapsed, and these were treated with doublet regimens. Hence, no conclusions can be drawn from this small series.
MMR deficient tumours were more frequent in the right colon (21.6 vs. 7.2%, p = 0.01), but did not influence DFS or OS. Even when stratifying by stage, the advantage of dMMR was not apparent in our analysis. The prognostic value of MMR has been evaluated in different studies. In a population-based study, MSI-H tumours had a better overall survival (HR 0.42, 95% CI 0.27-0.53, p < 0.001) and were associated with a decreased probability of metastasizing to regional lymph nodes (OR 0.49, 95% CI 0.21-0.53, p < 0.001) and distant organs (OR 0.49, 95% CI 0.27-0.89, p = 0.02) [20]. These tumours also exhibited a lower response to 5-FU, especially in stage II [18]. In our study, mutator phenotype was identified solely based on immunochemistry (IHC) of MMR proteins. Conflicting data have been published concerning the correlation between IHC and MSI [19, 21]. In the identification of Lynch syndrome patients, MSI testing (PCR) is considered as the reference tool although it is less accessible than IHC. Nevertheless, recent studies have shown that both techniques have high sensitivity and specificity in screening mutations in MMR genes [22, 23]. The major limitations of immunohistochemistry are due to rare missense mutations that are usually reported in MLH1 and MSH6 genes, which affect protein function. Therefore, IHC may fail to identify all the properties of the MSI phenotype [23]. These differences could account for some of the discrepancies found between the present and prior studies.
No germline mutations in MMR genes were identified so far in patients with dMMR tumours although this analysis is still not complete.
Rectal tumours without synchronous metastases at diagnosis had a higher rate of RAS mutation (60.7%), followed by MMR deficiency (10.7%). Only 1 case was BRAF mutated. In a recent retrospective study, rectal dMMR tumours had a significantly higher TNM downstaging after neoadjuvant chemoradiation, and DFS was significantly higher as compared to MMR proficiency tumours [24]. In our series, different molecular profiles did not correlate with different responses, although our numbers are too small to allow any definitive conclusions.
Right-sided tumours showed a trend towards a better DFS and OS in patients without metastases. Although previous studies suggest that right-sided tumours may have a worse prognosis with lower OS [1], the low rate of BRAF mutations in our series may have contributed to these discrepancies.
Our study has some limitations. We did not account for tumour heterogeneity, which can be highly variable, within primary tumours or between primary and metastatic sites [25]. In a recent systematic review and meta-analysis, metastatic CRC cancer showed high concordance with primary tumours, in multiple biomarkers, such as KRAS (93.7%), NRAS (100%) or BRAF (99.4%) [26]. Even though metastases usually contain genetic abnormalities observed in primary tumours, intra- or intertumour heterogeneity may occur, a phenomenon strongly evidenced in CRC tumours [26]. Phenotypic heterogeneity in tumours may constitute a major source of therapeutic resistance [27]. Therefore, treating metachronous metastasis based on the genetic profile of the primary tumour may not be completely appropriate. Finally, identification of tumour genotype was not possible in 170/206 patients with colonic tumours mainly related to difficulties in determining MMR status. Finally, our study had a relatively small sample size, which may justify the lack of significant results and discrepancies from the literature, namely in the prognosis of BRAF-mutated and dMMR tumours.
Although our study is much smaller than previously published clinical trials, it has the advantage of reflecting the potential benefit of studying tumour genotype in clinical practice. As of today, molecular testing is solely performed in clinical practice for the selection of patients with metastatic CRC for targeted biologic agents. In our retrospective study we observed that mutational analysis of CRCs may also allow important prognostication as RAS-mutated tumours have a lower DFS and OS thereby suggesting more intensive surveillance and/or more aggressive therapy, even in early stage II disease. Although our numbers are small, we also found that BRAF mutation only imprints a worse prognosis in relapsed patients, further supporting aggressive upfront therapy with FOLFIRINOX and bevacizumab after relapse, as previously recommended [18, 19]. Besides these potential advantages supporting molecular profiling of CRC we have to consider that, as of today, this analysis is time consuming, expensive and of little help in defining individualized therapies. Larger population studies are warranted to better understand the cost-effectiveness of this strategy when treating patients with CRC.

