










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkGE-Portuguese Journal of Gastroenterology
versão impressa ISSN 2341-4545
GE Port J Gastroenterol vol.22 no.6 Lisboa dez. 2015
https://doi.org/10.1016/j.jpge.2015.06.001
REVIEW ARTICLE
Systematic Review of the Relation Between Intestinal Microbiota and Toll-Like Receptors in the Metabolic Syndrome: What Do We Know So Far?
Revisão Sistemática da Relação Entre a Microbiota Intestinal e os Receptores Toll-Like na Síndrome Metabólica: O Que Sabemos Atualmente?
José Pedro Portela-Cidadea,∗, Marta Borges-Canhaa, Adelino Ferreira Leite-Moreiraa, Pedro Pimentel-Nunesa,b,c
a Physiology and Cardiothoracic Surgery Department, Cardiovascular Research & Development Unit, Faculty of Medicine, University of Porto, Porto, Portugal
b Gastroenterology Department, Instituto Português de Oncologia do Porto Francisco Gentil, Porto, Portugal
c CINTESIS/Biostatistics and Medical Informatics Department, Porto Faculty of Medicine, Porto, Portugal
* Corresponding author.
ABSTRACT
Introduction: Metabolic syndrome is an emerging problem in developed countries and presents itself as a potential threat worldwide. The role of diabetes, dyslipidaemia and hepatic steatosis as pivotal components of the metabolic syndrome is well known. However, their common persistent chronic inflammation and its potential cause still elude. This systematic review aims to present evidence of the mechanisms that link the intestinal microbioma, innate immunity and metabolic syndrome.
Methods: A comprehensive research was made using PubMed database and 35 articles were selected.
Results: We found that metabolic syndrome is associated to increased levels of innate immunity receptors, namely, Toll-like receptors, both in intestine and systemically and its polymorphisms may change the risk of metabolic syndrome development. Microbioma dysbiosis is also present in metabolic syndrome, with lower prevalence of Bacteroidetes and increased prevalence of Firmicutes populations. The data suggest that the link between intestinal microbiota and Tolllike receptors can negatively endanger the metabolic homeostasis.
Conclusion: Current evidence suggests that innate immunity and intestinal microbiota may be the hidden link in the metabolic syndrome development mechanisms. In the near future, this can be the key in the development of new prophylactic and therapeutic strategies to treat metabolic syndrome patients.
Keywords: Immunity, Innate; Intestinal Mucosa; Metabolic Syndrome X; Microbiota; Toll-Like Receptors
RESUMO
Introdução: A síndrome metabólica é, hoje, um problema emergente nos países desenvolvidos e apresenta-se como uma das principais ameaças médicas à escala global. O papel desempenhado pela diabetes, dislipidemia e a esteatose hepática, como componentes principais desta Síndrome é prontamente reconhecido. No entanto, a inflamação crónica persistente comum e as suas potenciais causas ainda não estão claramente definidas.
Objectivos: Esta revisão sistemática pretende apresentar evidências dos mecanismos que interligam o microbioma intestinal, a imunidade inata e a síndrome metabólica.
Métodos: Uma pesquisa sistemática foi realizada, utilizando a base de dados PubMed, tendo selecionado 35 artigos para a elaboração desta revisão.
Resultados: A síndrome metabólica está claramente associada a níveis aumentados de expressão dos receptores da imunidade inata, nomeadamente, os receptores da família Toll-like receptors, quer no tecido intestinal, quer sistemicamente, e diferentes polimorfismos parecem ser responsáveis por diferentes riscos de desenvolver esta doença. Por outro lado, a disbiose do microbioma intestinal está também presente na síndrome metabólica, com a presença de Bacteriodetes em menor prevalência e com aumento das populações de Firmicutes. Os resultados sugerem ainda que a ligação entre a microbiota intestinal e os receptores da imunidade inata possa negativamente comprometer a homeostasia metabólica, de forma semelhante à evidenciada nesta síndrome.
Conclusões: Evidência actual sugere e suporta que a imunidade inata e a microbiota intestinal possam ser a ligação pivô nos mecanismos de desenvolvimento da síndrome metabólica. Num futuro próximo, esta pode ser a chave para o desenvolvimento de novas estratégias profiláticas e terapêuticas para a síndrome metabólica.
Palavras-Chave: Imunidade Inata; Mucosa Intestinal; Síndrome X Metabólica; Microbiota; Receptores Toll-like
1. Introduction
The metabolic syndrome presents itself as one as the principal chronic diseases of the developed countries and an important determinant of cardiovascular and metabolic mortality risk.1 It is defined as a persistent pro-inflammatory state in which abnormal metabolic and physiological factors produce an increased risk of developing diabetes, obesity, dyslipidaemia and other cardiovascular risk factors.1-3 Recent data report a consistent activation of the innate immunity through the Toll-like receptors (TLR) and its downstream signalling, suggesting not only a potential causative way, but also a possible perpetuator of its chronic immune stress to the organism.4-6 On the other hand, new insights have revealed a pivotal role of the intestinal microbiota and its interaction with the host genetics, in the development of obesity and insulin resistance.7-9 It has been also described in the role of intestinal microbiota, its migration and its metabolic products systemic effects, in the activation of these TLR receptors in several organs, especially the liver.10,11
However, in spite of this new data, the relationship, causality and the mechanisms by which the intestinal microbiota can influence the expression of several immune receptors including TLR still eludes. Also, the relationship between differential expression of TLR and the lesion of several organs presented in metabolic syndrome is poorly understood.
This systematic review aims to access the most recent data about the relevance of intestinal microbiota and TLR expression in the development of hepatic lesion and metabolic syndrome.
2. Methods
A comprehensive search was performed in PubMed and the following queries were used: ((Metabolic Syndrome(All Fields) AND (microbiome(All Fields) OR microbiota(All Fields))) OR (Metabolic Syndrome(All Fields) AND (Tolllike receptors(All Fields) OR TLRs(All Fields)) AND (microbiome(All Fields) OR microbiota(All Fields)))),and ((gut microbiota(Title) OR microbiota(Title)) AND (((TLR(Title)) OR Toll like receptor(Title)) OR Innate immunity(Title))).
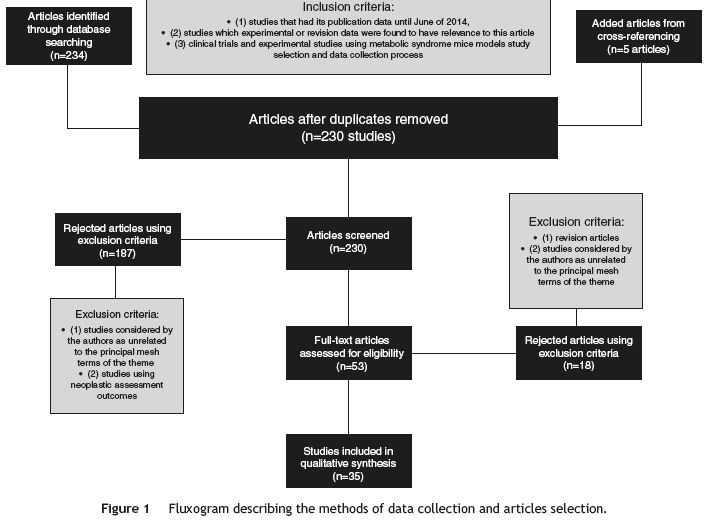
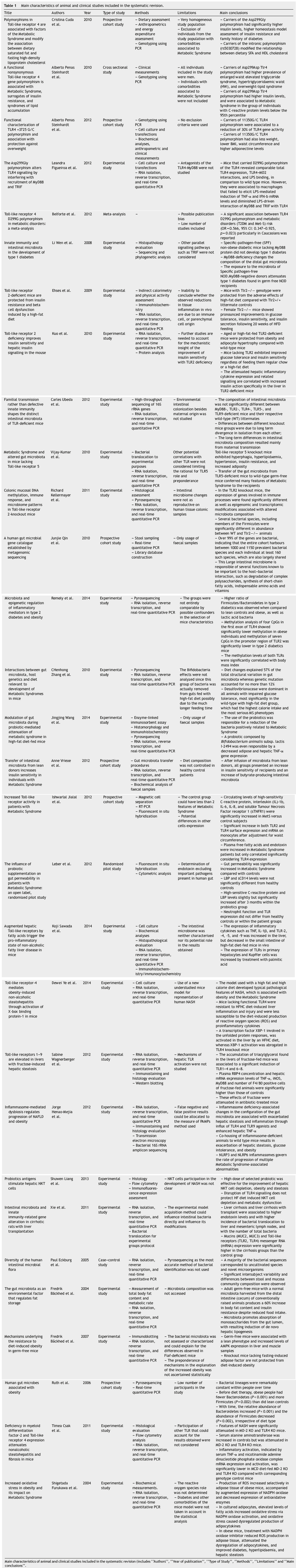
From the search and after duplicates were removed, 230 studies were retrieved (Fig. 1). Using inclusion and exclusion criteria, 35 articles were selected, analyzed and included in this revision (Table 1).
Figure 1
Table 1
The principal summary measures in an outcome level were different risk ratios of developing obesity, diabetes, dyslipidaemia or metabolic syndrome in every experimental conditions used, difference in means of RNA or biochemical expression levels of markers studied and differences on histological or phenotypical assessment of major organs studied n a variety of experimental conditions. Subgroup analyses within the selected studies were also considered and taken into account.
3. TLR expression and metabolic syndrome: the importance of the innate immunity
It is well known that the TLRs comprehend an extended family of pathogen-associated molecular patterns recognition (PAMP) receptors of the innate immunity. These receptors allow the prompt activation of downstream signalling of MyD88-dependent and MyD88-independent pathways in response to several antigens, mainly of Gram-positive and Gram-negative bacteria, and they are all linked to production of cytokines and enhancement of the inflammation.
Since it is established that subclinical inflammatory processes are major contributors to metabolic syndrome, obesity and diabetes, several lines of investigation were made to confirm the role of innate immunity receptors and prompt activation in the pathogenesis of metabolic disturbances.12
An initial study shows that there was a significant increase in both TLR2 and TLR4 cell surface expression and mRNA levels in monocytes of metabolic syndrome patients. This phenomenon persisted even when its values were statistically adjusted to waist circumference and body mass index. This study also reveals that this TLR increased activity was significantly correlated with significant higher blood pressure and plasma glucose levels, and with interleukin-1b (IL-1b), Monocyte Chemoattractant Protein-1 (MCP-1) and Necrosis Factor-kB (NF-kB) activity, concluding that the activation of the TLR can count as an independent factor not only to metabolic syndrome but also to the cardiovascular risk of these patients.13
The impact of TLRs is also verified in other clinical situations that are strongly related to metabolic syndrome and that can explain its participation as a causative factor through indirect pathways.
On one hand, Cristina Cuda et al showed that different polymorphisms of TLR4, with different activation rates of this receptor, were statistically associated to different serum insulin levels and insulin sensitivities. The individuals with TLR4 with 299Gly allele were found to have higher levels of insulin and lower levels of insulin sensitivity in comparison to the considered normal Asp homozygotes. They also found that a second polymorphism, an intronic SNP (rs5030728) can even modulate the relationship between dietary saturated fatty acids and HDL cholesterol, showing that an increased intake of saturated fatty acids was inversely related to significant lower levels of HDL cholesterol, in these individuals.14 Steinhardt et al,15 on the other hand, strongly confirmed that another polymorphism of TLR4 is associated to syndromes of lipid accumulation, thereby sustaining that TLR could be involved in the regulation of several metabolically substrates and increase the risk of dyslipidaemia, a known factor related to metabolic syndrome.
Another line of evidence using mice with +3725G/C polymorphism of TLR4, shows that these mice have a functional decrease in their receptor activity to only 30% (p = 0.0001), and that they presented a statistically significant association with lower body mass indexes (25.5363.50 vs. 28.6064.62 kg/m2; age adjusted p = 0.023), waist circumferences ((89.27614.46 vs. 97.51612.59 cm; age adjusted p = 0.025), and with higher adiponectine levels (14.5 ng/mL vs. 10.6 ng/mL; p = 0.021).16 These results may denote a positive relation between a prompt activation of the innate immunity and obesity, a known factor present in Metabolic Syndrome patients. These remarkable results, however, were not so strongly verified in the smoking population, and the authors point this factor as a potential confounding variable that must be considered in future studies.
A strong association has been also described between the expression and activity of TLRs and the development of type 1 and type 2 diabetes, major factors in the metabolic syndrome pathogenesis.17
A recent study described that Tlr2−/− genotype mice after 12 weeks of high-fat diet not only had more glucose tolerance and insulin sensitivity than the control group, but also presented reduced levels of leptin, MCP-1 and tumour necrosis factor-a (TNF-a). Interestingly, insulin sensitivity in TLR2 knockout mice was conserved in the liver causing a significant statistical reduction of hepatic glucose production and lower risk of developing hepatosteatosis under high-fat diet.18 These data were consistent with Kuo et al. study that found that TLR2 knockout mice presented lower glucose and insulin levels than their wild counterpart, revealing an important role of this receptor in glucose homeostasis. This same study also stated that mice that knockout to TLR2 were associated to a phenotype of decreased body weight, fat mass, lower number (1.21×107 in Tlr2−/− vs. 2.62×107 in WT) and smaller adipocyte cells (mean area 821mm2 in Tlr2−/− vs. 1503mm2 in WT).19
A meta-analysis using studies that linked type 2 diabetes, metabolic syndrome and D299G polymorphism of TLR4 also showed that this miss-sense variant was responsible for a lower response of this receptor to its ligand, lipopolysaccharide, probably through a deficient recruitment of its down-signalling adapters MyD88 and TIR-domain-containing adapter-inducing interferon-b (TRIF). The study concluded that the D299G-variant was, in the Caucasian subgroup, associated to a reduced risk of diabetes type 2 and metabolic syndrome. However the results were not reproducible in other subgroups and the authors stated that it could be explained by the differences in the allele frequency among different populations.17,20
4. Microbiota and metabolic syndrome: a new link
Recently, the intestinal microbiota has been progressively studied and knowledge of its profound influence in maintaining the human physiology and nutrition has astonishingly increased.21 Using recent methods of pyrosequencing, the human gut microbial population has been described as the one with the highest in what concerns to density and variability of organisms, comprehending more than 5000 bacterial taxa and composed mainly by Bacteroidetes and Firmicutes among other phyla that are less prevalent such as Actinobacteria, Proteobacteria, Fusobacteria, Deferribacteres and Deinococcus.22 Although the preponderant presence of these microorganisms in our gut, their impact in the human homeostasis is still not fully known and its relationships with several chronic diseases, especially with the Metabolic Syndrome, has opened new lines of pathological evidence and potential therapeutic targets.
In fact, it has become apparent that the microbiota can change metabolically the way we acquire nutrients or our ability to harvest energy through several mechanisms.23 The preponderance of these bacteria in the catalytic processing of carbohydrates in the human digestion or even the precise location where these metabolic reactions occur can influence their availability to the human organism.23 The bacteria are also associated to functions such as stabilizing tight junctions and promoting the secretion of anti-inflammatory cytokines.23 In the lipid metabolism, several types of conjugated and free fatty acids are generated by the intestinal microbiota and play important roles in stimulating paracrine and endocrine peptides such as glucagon-like peptide (GLP) and peptide YY, modulating the intestine function.24
Interestingly, two studies by Backhed et al. compared mice raised in the absence of any microorganisms (germfree), with mice in which was introduced a microbiota from the cecum of other conventional mice raised in non-germ free conditions, through a process that the authors called conventionalization. Not only were the germ free mice found to be resistant to diet-induced obesity even in low-fat polysaccharide-rich diet, but also these conventionalized mice had 42% more total body fat, which represented a dramatic increase of 57% in their total body fat content and a 61% increase in epididymal fat weight, in spite of the Western diet and reduced chow consumption. The authors also verified that these mice had statistical elevations of liver mRNAs encoding enzymes that were participants in the fatty acid biosynthetic pathway, leading to an increase of triglycerides production in their livers.23,24
These facts point to influences on different energetic inputs, nutrients availability and metabolic modulations by intestinal microbiota, showing an important contribution to metabolic disturbances similar to those described in obesity and diabetes.
Furthermore, it has become apparent that in spite of its location in the human intestine, the microbiota plays important systemic changes that are directly linked to the low-grade chronic inflammatory state, hugely present in the metabolic syndrome.
On one hand, the characterization of the intestinal bacteria in obese model mice (ob/ob) was consistent with 50% reduction of Bacteroidetes abundance and a reverse increase of Firmicutes when compared to lean mice.25 This unbalanced microbiota was found to be modulated by the composition of the diet, mainly rich in fatty acids, independent of previous obesity or obese state, since resistant models to high-fat induced obesity mice did experience the same microbiome changes that did its wild counterpart.25 Moreover, it was found that a high-fat diet simulated the infusion of bacterial LPS defining a phenotype with development of obesity, diabetes and inflammatory cells infiltration in the adipose tissue. It was also found that the gut bacteria were able to induce the suppression of fastinginduced adipocyte factor, thereby limiting the inhibition of Lipoprotein lipase in the adipocytes and promoting the body fat accumulation.6
Analogous results were obtained by Remely et al study, since higher prevalence of Firmicutes and lactic bacteria were encountered in type 2 diabetic mice in opposition to Bacteroidetes and F. prausnitzii. The authors also tested the influence of these population changes in the methylation of exons of TLR2 and 4 in the intestinal epithelium and described significant statistical lower methylations rates for TLR2 in diabetic mice and lower methylation rates for TL4 in obese mice, suggesting that there are important epigenetic modulations by the intestinal bacteria and that those modulations could probably incur in exacerbated risks of diabetes and obesity.26
These results support the previous data from Zhang et al study that showed reduced levels of gut-protecting Bifidobacteria, a phylum associated with resistance to the effects of LPS from the gut microbiota, in mice with a high-calorie diet. The authors suggest that through the modification of intestinal microbiota to a pathogenic-like microbiome, a high-calorie diet can be responsible for a loss of integrity in the barrier function of the intestine, the systemic spread of bacterial endotoxins and pro-inflammatory state. In fact the study points that a high inflammatory state was present in all animals with impaired glucose tolerance and increased body fat, on high-fat diet. The authors also revealed a predominance of Desulfovibrionaceae, sulphatereducing bacteria, in high-calorie diet mice, a fact that could be related to the development of metabolic syndrome in these mice, since the disruptive properties of the sulphate in the intestinal barrier were present. Finally, the authors described that this transformation to a pathogenlike population through diet, seems to play a relevant role in developing metabolic syndrome that is, apparently, also independent of the host genetics and genomic expression.27
Backhed et al aimed to test the importance of microbiota during infancy in the development of obesity later in life. This study showed that breastfeeding seems responsible for the colonization of the intestine mainly by Bifidobacteria and for a decrease in the Enterobacteriaceae family members. The variations to this intestinal microbiome ecosystem, with reduced levels of protective strains such as the Bifidobacteria, that are observed in 2-year-old infants, had a profound impact on a regulator of the lipoprotein lipase called Angpt14/Fiaf, which seems to act as an inhibitor of the enzyme activity and is responsible for an increase in the body fat accumulation. This was observed in studies using mice models, which showed an increase in body fat tissue not only in germ-free mice with Angpt14/Fiafdeficiency, but also in non-germ-free mice, with a functional Angpt14/Fiaf protein.
Also, the study shows that the use of antibiotics in infants can cause the elimination of Bacteroids and a reduction in Bifidobacterium population levels, thereby reducing two important antiobesogenic families in the intestinal microbiota.23
5. TLR, mycrobiota and Metabolic Syndrome: a global vision
The activation of innate immunity through the TLR cannot be dissociated from the intestinal microbiota and its mutual crosstalk playing a decisive factor in the development of metabolic syndrome.
In fact, the modulation and interaction between microbiota and TLR activation were investigated in a TLR2 knockout mice study that revealed that, in spite of the genetic predetermination to insulin sensitivity by these mice reported in studies before,18,19 this phenotype could be drastically reversed by gut microbiota. The study confirmed that these TLR2 knockout mice, in non-germ-free facilities, had higher levels of Firmicutes and Bacteroidetes in comparison to wild-type mice, and increased lipopolysaccharides absorption, glucose intolerance, insulin resistance and obesity. The authors stated that these results can be explained by an increased activation of TLR4 receptors in the absence of TLR2 activation. They also verified increased endoplasmic reticulum stress and JNK activation by the TLR4, which had effects that could be inhibited by a TLR4 antisense oligonucleotide causing an increase in glucose tolerance and sensitivity when compared to the controls. Furthermore, in TLR2 knockout mice, the use of a mixture of antibiotics resulted in an improvement of insulin sensitivity and metabolic status.7
Another recent study by Ubeda et al further revealed that using knock-out mice to a specific TLR, the impact of the absence of that certain TLR was initially minimal in the bacteria communities at the intestine, after a treatment with antibiotics, when compared to their respective wild-type mice. This evidences that some caution is advised when valorizing microbial communities differences in these groups. Interestingly, the authors also stated that differences in mices colonies with different TLR deficiencies were only verified after long-term breeding in isolation from each other, and probably with great influence from maternal transmission.28 However, contrary to the previous study of Vijay-Kumar et al29 that described a major link between TLR-5 KO mice and the development of metabolic syndrome, this study did not find any difference in the intestinal microbiota colonies or in the risk of metabolic syndrome. The authors attribute these results to differential exposures in husbandry conditions and mice types.28
To add to this complex network of relations between host genetic expression and the environment, Kellermayer et al. study showed an increased risk of epigenetic modification, DNA methylation and differential rates of transcripts in TLR2-knockout mice in the intestinal mucosa epithelium. The authors pointed out that these epigenetics and metagenomic effects, along with the modulation of the bacterial populations, could explain the loss of the protective barrier function of the intestine and changes in the microbiome environment observed in the Metabolic Syndrome.30
As the importance of the intestinal microbiome in the metabolic syndrome becomes more apparent, new lines of investigation have appeared, aiming the bacterial populations as a therapeutic hypothesis in the Metabolic Syndrome.
A study by Wang et al used high-fat diet mice in which was induced metabolic syndrome and subjected them to administration of one of three probiotics (Lactobacillus paracasei, L. rhamnosus and Bifidobacterium animalis) in order to modulate the gut microbiota. The results showed that these strains were able to reduce the weight gain, increase insulin sensitivity and limit hepatic steatosis, and also that they were able to modulate innate immunity and the pro-inflammatory state, through the reduced infiltration of macrophages in the adipose tissue. Moreover, the probiotic treatment was capable of reducing key phylotypes keen to cause metabolic syndrome (as are Desulfovibrionaceae and Clostridium examples), and capable of reducing inflammatory processes associated to sulphate sodium-induced lesion by these bacteria. On the other hand, certain strains and also certain combinations of probiotic strains can probably provide different benefits in reversing the metabolic syndrome by a different impact on certain mechanisms, such as the increase of acetate, a known molecule shown to be able to improve obesity and diabetes on these mice.31
Another study, performed in humans, reported that infusion of intestinal microbiota collected from lean donors into the intestine of Metabolic Syndrome subjects was able to increase insulin sensitivity (median rate of glucose disappearance changed from 26.2 to 45.3 mol/kg/min; p < 0.05), after 6 weeks. These changes were associated to an increase of butyrate-producing bacteria in the guts recipients, which is known to influence directly the glucose metabolism.32 Another study using probiotics rich in Lactobacillus verified that those who received the probiotics had lower body mass index, waist and hip measurements, and presented a reduction of fat in the abdomen and subcutaneous areas (p < 0.01).32
A randomized pilot study by Leber et al. tried to unravel the effect of a Lactobacillus probiotic strain in the gut permeability and bacterial endotoxin presence, in human metabolic syndrome patients, covering two phenomena attributed to the pathogenesis of the syndrome. However, in spite of the significant increase of the gut permeability in the metabolic syndrome group of patients, the investigators did not find any difference of the endotoxin, C-reactive protein, neutrophil function and TLR expression levels between the group who had received the probiotic and the control group.33
6. Liver as a pivotal organ in metabolic syndrome
The liver is a major organ of the human organism and it has a profound impact in its metabolic and immune homeostasis.10 Indeed, its high efficient metabolic regulation of glucose, fatty acids and immune regulators and its privileged location as an organ that first receives blood from the gastrointestinal tract, protecting the remaining circulation from its potential threats, makes the liver a crucial participant in several regulatory processes. Furthermore it has been progressively verified that these same functions can be unbalanced in the metabolic syndrome. In fact, a prompt activation of several immune receptors on the hepatic immune system occurs in response to a break of the intestinal epithelial barrier function and to an increase of certain bacterial populations and their translocation through the vascular system.10
Therefore, this immune mechanism to disrupted intestinal homeostasis can explain the development of non-alcoholic fatty liver disease the development of several comorbidities such as diabetes, dyslipidaemia and obesity, and its increased risk to metabolic syndrome.1
Sawada et al aimed to measure the expression of TLRs, TNF-a, interleukin-1B and phospho-interleukin-1 receptor-associated kinase 1, in the liver and small intestine, in non-alcoholic fatty liver disease induced mice. The authors reported a significant statistical increase in the inflammatory cytokines and TLR2, 4, 5 and 9 expressions in the liver of 16-week mice, but no differences in the liver of 4- and 8-week mice. The study also revealed that, in the intestine, exactly the opposite could be observed, since all parameters were significantly decreased indicating that these pathways are probably inhibited in mice with NAFLD. Moreover, the treatment of these mice with antibiotics could attenuate the higher expression of those inflammatory markers and TLRs in the liver but it had little impact on the expression of these molecules in the small intestine of NAFLD mice.34
The authors further stated that, the increase on the liver TLRs expression appears to be selective since TLR2, 4, 5 and 9 were upregulated in primary Kupffer cells while only TLR4 and 9 were increasingly expressed in the primary hepatocytes.34
Another recent study tested the effect of TLR4 in NASH in high-fat, high-cholesterol diet mice. The results stated that, compared with wild-type, the TLR4 mutated mice were resistant to the development of macrovesicular and microvesicular steatosis, hepatocellular ballooning and a fivefold increase in NASH scores. This phenotype was even associated to a decreased TLR4 function mainly in Kupffer cells, and consequential lower levels of macrophages and cytokines. Interestingly, the study also included the measurement of a transcription factor named XBP-1, known to be stimulated by reactive oxygen species and a part of the responsive pathway to that oxidative stress through the unfolded protein response cascade. This protein was abrogated in the TLR4 mutant mice, with a simultaneously decrease of NF-kB activation and cytokine production.35
Also, TLR4 has been described as a signalling way to NAFLD development, in high-fat diet mice, and linked to hepatic steatosis, hepatic insulin resistance and hepatic weight gain in these experimental circumstances. The same results were found through other members of TLR family such as TLR9 and TLR5 pathways.36 Finally TLR2 was also studied and is now apparent that its ligands are increased in obese mice and that its blockage can restrain its insulin resistance induction. In fact, TLR2 knockout mice exhibit lower expression of TNF-a and IL-1b inflammatory cytokines.19
The same rational was presented in other studies, where fructose-induced hepatic steatosis in mice was associated with significant induction of TLR1-4 and TLR6-8 along with an increase of MyD88, TNF- and iNOS levels. The authors point out that bacterial components translocation and the prompt activation of TLRs and TLR-dependent pathways support a chronic inflammatory state that could, in the liver, end with the development of non-alcoholic liver disease.29,33,37
Further studying the participation of the inflammasome pathway in the NAFLD pathology, Henao-Mejia et al tested two inflammasome molecules and its effector protein in bacterial modulation and metabolic syndrome outcome. The study revealed that the progress from NAFLD to NASH was positively influenced by these molecules since knockout mice to inflammasome proteins (Nlrp3 and Nlrp6) were associated to lower effector protein (IL-18) levels, different gut microbiota and increased TLR4 and TLR9 agonists. These mice also had more hepatic steatosis, obesity and glucose intolerance and higher rate of Porphyromonadaceae in the intestinal gut, a known bacterial family that increases the risk of metabolic syndrome in both mice and humans.38
Considering its importance, a modulation by probiotics can emerge as a therapeutic target to this metabolic infection. A recent study by Liang et al. using probiotics showed an improvement in the obesity, glucose tolerance and insulin levels in high-fat diet-fed mice. It also revealed that hepatic steatosis was reduced with significantly lower hepatic triglyceride content and improved histology, with the use of this probiotics.39
Another study by Xie et al used normal and liver cirrhosis mice that were subjected to liver transplantation. The authors found that not only the liver cirrhosis groups had higher endotoxin levels and higher numbers of total bacteria, but also, the referred group had an increased number of bacteria in liver and lymph nodes. The liver cirrhosis groups had higher rates of Enterobacteriaceae and lower Lactobacilli and Bacteroides and, simultaneously, had higher MUC2, MUC3 and TLR2 and 4 mRNA expression levels in the intestine epithelium, and TLR2 and 4 increased mRNA expression levels in the liver. Astonishingly, these parameters had not improved until 1 month after the liver transplantation and, surprisingly, had not improved with the use of a lactobacillus-enriched probiotic, suggesting probably the need of longer times of therapy or search for different probiotic compositions specific for each metabolic level of disturbance, either hepatic or systemic.40 This rational may account for important medical decisions in the future and possibly allow different therapeutical options become medically used in clinical practice.
7. Discussion
For all the data stated, it is well apparent that the interaction between intestinal microbiota and the innate immunity might have a pivotal role in the metabolic syndrome, which had impact that could go way beyond the limits of the intestine and have a direct and systemic impact through several organs (Table 2).
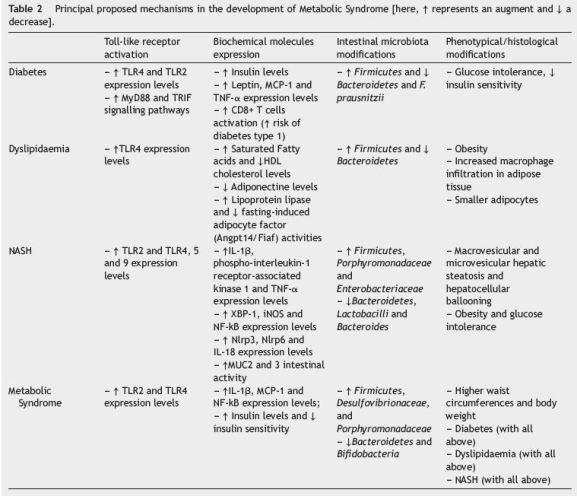
Based on the results analyzed, we propose that the composition of the intestinal microbiome, changing from a symbiotic population mainly composed by Bifidobacteria and Bacteroidetes, to a more unbalanced one, where Firmicutes, Porphyromonadaceae and Desulfovibrionaceae emerge, can be modulated and provoked by diet and its relative composition in lipids and other factors. This dysbiotic environment can promptly activate several immune receptors present in the intestinal barrier though the recognition of PAMPs and bacterial antigens, culminating in an increased activation of TLR2, 4, 5 and 9, and other synergic inflammatory signalling pathways that were not described yet. The local pro-inflammatory profile can explain the altered phenotypic expression of these receptors at the intestine surface, the metabolic absorption disturbances observed, the increase of cytokines and inflammatory markers levels and the loss of barrier function of the intestine, which are all described in metabolic syndrome models.41,42
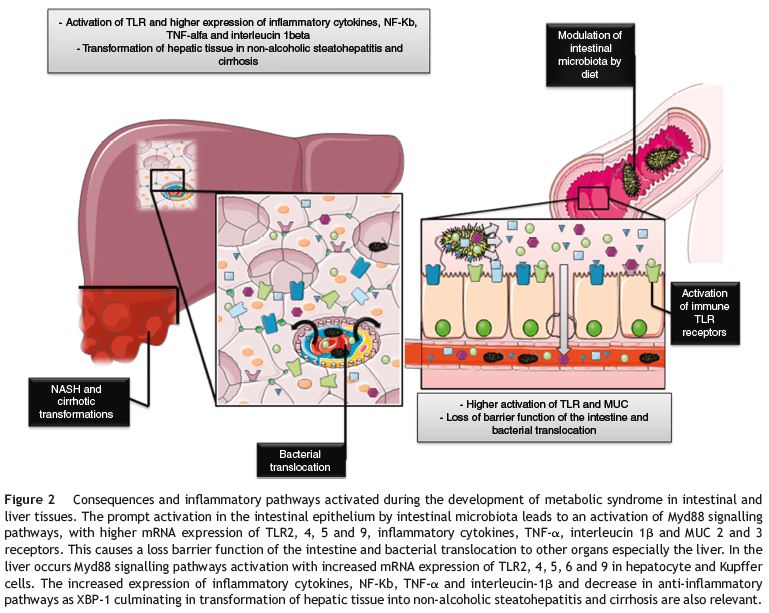
With this break of intestine mechanic defences, this dysbiotic bacteria and its products will have an easy access to the vascular space, raising the level of exogenous provocative antigens to the immune innate system and through its translocation influence the homeostasis of different organs, specially the liver because of its metabolic preponderance and privileged location43 (Fig. 2).
Figure 2
These mechanisms synergistically account for the increased activation of TLR2, 4, 5 and 9 in the Kupffer cells and the TLR2, 4 and 9 in the hepatocytes and the consequential increase in the inflammatory products of its pathways such as TNF-a, IL-1b and iNOS. This chronic inflammatory state can justify the appearance of macrovesicular and microvesicular steatosis, and hepatocellular ballooning that counts for the NASH transformation.44 The role and overexpression of several other molecules as xBP1 and NFKb can further explain this modification probably through a shift in the genetic expression and/or epigenetic modulation of the liver cells.
On the other hand, throughout the organism, the antigenic activation of TLR2 and TLR4 in various cells can be a major way of explaining the glucose intolerance, insulin resistance and the increased risk of diabetes, and comorbidity associated to this syndrome. Furthermore, the MyD88 pathway activated by these receptors seems to be preponderant to this outcome, since the absence of its signalling participants or TRIF could account for a reduced risk of diabetes developed.5
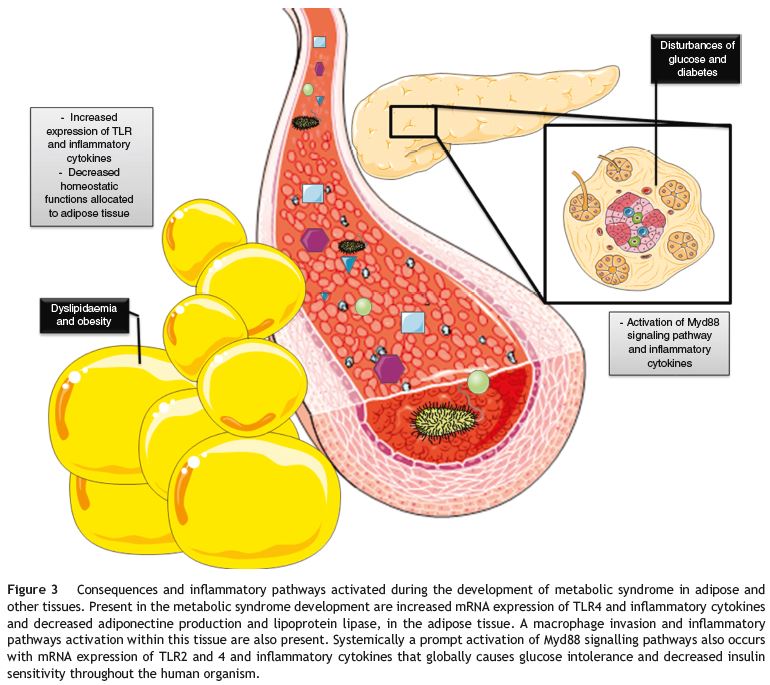
The innate immunity activation can even influence the milieu of the adipose tissue, being responsible for a pro-inflammatory state with increased activation of TLR4, the recruitment of different inflammatory cells and the production of inflammatory molecules. This can be the cause of the decreased adiponectine levels expression, the increased lipoprotein lipase functions and the explanation of the dyslipidaemia and obesity present in these patients45 (Fig. 3).
Figure 3
Furthermore, the chronic inflammatory state is known to disturb the balance of coagulation/anticoagulation factors and the metabolic production of oxidative stress throughout the organism. These factors can further influence the regulation of delivering nutrients and energetic substrates to the cells and its internal management, causing a most profound impact in the homeostatic equilibrium of the human body.46
These new data open interesting lines of evidence that a modulation of intestinal microbiota can be a powerful therapeutic way of influence or even reverse the progression of metabolic syndrome. The use of probiotics has revealed that through changes in the intestinal dysbiosis, a lower pro-inflammatory profile can be achieved with expressive benefits in glucose tolerance, insulin sensitivity, lower weight and lower hepatic steatosis. Although several lines of investigation are still needed on this behalf, the data collected so far are consistent with a potential therapeutic target in the clinical control of metabolic syndrome development and treatment.47
In conclusion, this review detects several lines of evidence of the profound preponderance of early activation of TLR receptors and its interactions with a dysbiotic intestinal microbiota in causing several disturbances known to be implicated in metabolic syndrome. Thus, in the near future, the understanding if there is a specific set of TLR over-expression associated to the different chronic metabolic diseases that coexist in the metabolic syndrome, the characterization of the modulation of hepatic immune receptors and cytokine production and the clarification of which are the major bacterial molecules that work as a factor to propagate or ameliorate the metabolic syndrome state, shall be preponderant points in the breakthrough of the investigation in metabolic syndrome medical area.
References
1. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162. [ Links ]
2. Tilg H. Obesity, metabolic syndrome, and microbiota: multiple interactions. J Clin Gastroenterol. 2010;44:S16-8. [ Links ]
3. Mehal WZ. The Gordian Knot of dysbiosis, obesity and NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10:637-44. [ Links ]
4. Jialal I, Kaur H, Devaraj S. Toll-like receptor status in obesity and metabolic syndrome: a translational perspective. J Clin Endocrinol Metab. 2014;99:39-48. [ Links ]
5. Jin C, Henao-Mejia J, Flavell RA. Innate immune receptors: key regulators of metabolic disease progression. Cell Metab. 2013;17:873-82. [ Links ]
6. Parekh PJ, Arusi E, Vinik AI, Johnson DA. The role and influence of gut microbiota in pathogenesis and management of obesity and metabolic syndrome. Front Endocrinol (Lausanne). 2014;5:47. [ Links ]
7. Caricilli AM, Picardi PK, de Abreu LL, Ueno M, Prada PO, Ropelle ER, et al. Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol. 2011;9:e1001212. [ Links ]
8. Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol. 2014;60:197-209. [ Links ]
9. Frasinariu OE, Ceccarelli S, Alisi A, Moraru E, Nobili V. Gut-liver axis and fibrosis in nonalcoholic fatty liver disease: an input for novel therapies. Dig Liver Dis. 2013;45:543-51. [ Links ]
10. Seki E, Schnabl B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J Physiol. 2012;590:447-58. [ Links ]
11. Chassaing B, Etienne-Mesmin L, Gewirtz AT. Microbiota-liver axis in hepatic disease. Hepatology. 2014;59:328-39. [ Links ]
12. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752-61. [ Links ]
13. Jialal I, Huet BA, Kaur H, Chien A, Devaraj S. Increased toll-like receptor activity in patients with metabolic syndrome. Diabetes Care. 2012;35:900-4. [ Links ]
14. Cuda C, Badawi A, Karmali M, El-Sohemy A. Polymorphisms in Toll-like receptor 4 are associated with factors of the metabolic syndrome and modify the association between dietary saturated fat and fasting high-density lipoprotein cholesterol. Metabolism. 2011;60:1131-5. [ Links ]
15. Steinhardt AP, Aranguren F, Tellechea ML, Gómez Rosso LA, Brites FD, Martínez-Larrad MT, et al. A functional nonsynonymous toll-like receptor 4 gene polymorphism is associated with metabolic syndrome, surrogates of insulin resistance, and syndromes of lipid accumulation. Metabolism. 2010;59:711-7. [ Links ]
16. Penas-Steinhardt A, Barcos LS, Belforte FS, de Sereday M, Vilariño J, Gonzalez CD, et al. Functional characterization of TLR4 +3725 G/C polymorphism and association with protection against overweight. PLoS ONE. 2012;7:e50992. [ Links ]
17. Belforte FS, Coluccio Leskow F, Poskus E, Penas Steinhardt A. Toll-like receptor 4 D299G polymorphism in metabolic disorders: a meta-analysis. Mol Biol Rep. 2013;40:3015-20. [ Links ]
18. Ehses JA, Meier DT, Wueest S, Rytka J, Boller S, Wielinga PY, et al. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a highfat diet. Diabetologia. 2010;53:1795-806. [ Links ]
19. Kuo LH, Tsai PJ, Jiang MJ, Chuang YL, Yu L, Lai KT, et al. Toll-like receptor 2 deficiency improves insulin sensitivity and hepatic insulin signalling in the mouse. Diabetologia. 2011;54:168-79. [ Links ]
20. Figueroa L, Xiong Y, Song C, Piao W, Vogel SN, Medvedev AE. The Asp299Gly polymorphism alters TLR4 signaling by interfering with recruitment of MyD88 and TRIF. J Immunol. 2012;188:4506-15. [ Links ]
21. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59-65. [ Links ]
22. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635-8. [ Links ]
23. Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718-23. [ Links ]
24. Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104:979-84. [ Links ]
25. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022-3. [ Links ]
26. Remely M, Aumueller E, Jahn D, Hippe B, Brath H, Haslberger AG. Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benef Microbes. 2014;5:33-43. [ Links ]
27. Zhang C, Zhang M, Wang S, Han R, Cao Y, Hua W, et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 2010;4:232-41. [ Links ]
28. Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, et al. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med. 2012;209:1445-56. [ Links ]
29. Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228-31. [ Links ]
30. Kellermayer R, Dowd SE, Harris RA, Balasa A, Schaible TD, Wolcott RD, et al. Colonic mucosal DNA methylation, immune response, and microbiome patterns in Toll-like receptor 2-knockout mice. FASEB J. 2011;25:1449-60. [ Links ]
31. Wang J, Tang H, Zhang C, Zhao Y, Derrien M, Rocher E, et al. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J. 2015;9:1-15. [ Links ]
32. Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913-6, e917. [ Links ]
33. Leber B, Tripolt NJ, Blattl D, Eder M, Wascher TC, Pieber TR, et al. The influence of probiotic supplementation on gut permeability in patients with metabolic syndrome: an open label, randomized pilot study. Eur J Clin Nutr. 2012;66:1110-5. [ Links ]
34. Sawada K, Ohtake T, Hasebe T, Abe M, Tanaka H, Ikuta K, et al. Augmented hepatic Toll-like receptors by fatty acids trigger the pro-inflammatory state of non-alcoholic fatty liver disease in mice. Hepatol Res. 2014;44:920-34. [ Links ]
35. Ye D, Li FY, Lam KS, Li H, Jia W, Wang Y. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut. 2012;61:1058-67. [ Links ]
36. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47:571-9. [ Links ]
37. Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D, et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. 2011;300:G433-41. [ Links ]
38. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179-85. [ Links ]
39. Liang S, Webb T, Li Z. Probiotic antigens stimulate hepatic natural killer T cells. Immunology. 2014;141:203-10. [ Links ]
40. Xie YR, Liu SL, Liu X, Luo ZB, Zhu B, Li ZF, et al. Intestinal microbiota and innate immunity-related gene alteration in cirrhotic rats with liver transplantation. Transplant Proc. 2011;43:3973-9. [ Links ]
41. Henao-Mejia J, Elinav E, Thaiss CA, Flavell RA. The intestinal microbiota in chronic liver disease. Adv Immunol. 2013;117:73-97. [ Links ]
42. Sommer P, Sweeney G. Functional and mechanistic integration of infection and the metabolic syndrome. Korean Diabetes J. 2010;34:71-6. [ Links ]
43. Burcelin R, Garidou L, Pomie C. Immuno-microbiota cross and talk: the new paradigm of metabolic diseases. Semin Immunol. 2012;24:67-74. [ Links ]
44. Moschen AR, Kaser S, Tilg H. Non-alcoholic steatohepatitis: a microbiota-driven disease. Trends Endocrinol Metab. 2013;24:537-45. [ Links ]
45. Duseja A, Chawla YK. Obesity and NAFLD: the role of bacteria and microbiota. Clin Liver Dis. 2014;18:59-71. [ Links ]
46. Manco M, Putignani L, Bottazzo GF. Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev. 2010;31:817-44. [ Links ]
47. Miura K, Ohnishi H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:7381-91. [ Links ]
* Corresponding author.
E-mail address: zencidade@gmail.com (J.P. Portela-Cidade).
Ethical disclosures
Protection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of data. The authors declare that no patient data appear in this article.
Right to privacy and informed consent. The authors declare that no patient data appear in this article.
Conflicts of interest
The authors have no conflicts of interest to declare.
Received 14 May 2015; accepted 17 June 2015


{kind=link}
{kind=link}
{kind=link}
{kind=link}