










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Medicina Geral e Familiar
versão impressa ISSN 2182-5173
Rev Port Med Geral Fam vol.34 no.5 Lisboa out. 2018
https://doi.org/10.32385/rpmgf.v34i5.11945
RELATOS DE CASO
Polineuropatia amiloide familiar de início tardio: o desafio do diagnóstico
Late-onset familial amyloid polyneuropathy: a challenging diagnosis
Andreia Sofia Ferreira Marques,* Marisa Daniela Correia Garcês*
*Médicas Internas de Medicina Geral e Familiar. USF São João de Sobrado.
Endereço para correspondência | Dirección para correspondencia | Correspondence
RESUMO
Introdução: A polineuropatia amiloide familiar (PAF) é uma amiloidose hereditária que se manifesta através de uma neuropatia mista (sensitiva, autonómica e motora) de caráter progressivo. Este caso clínico evidencia a importância do médico de família no diagnóstico desta patologia, enquanto realça as principais dificuldades nesse diagnóstico em idades mais tardias e quando não existe história familiar conhecida.
Descrição do caso: Mulher de 71 anos, com antecedentes pessoais de carcinoma ductal in situ, síndroma do túnel cárpico e bócio multinodular adenomatoso. Nos últimos cinco anos apresentou múltiplas queixas inespecíficas, como obstipação crónica, dores nevrálgicas e parestesias das mãos e pés, perda progressiva de controlo do esfíncter urinário, deterioração progressiva da capacidade de locomoção, perda de peso. Não havia história familiar de PAF, exceto um irmão de 65 anos com diagnóstico de PAF de início tardio, após apresentar queixas de parestesias das mãos e pés. Esta informação alertou para a hipótese de diagnóstico de PAF, capaz de integrar as suas múltiplas queixas, a qual se veio a comprovar após teste genético.
Comentário: Este caso clínico apresenta uma série de particularidades que nos devem alertar para o facto de esta patologia poder surgir com um padrão diferente do que foi descrito por Corino de Andrade: a idade de início, a ausência de progenitores afetados, bem como o atingimento de famílias sem naturalidade na Póvoa de Varzim. Assim, o conhecimento de variantes tardias da PAF, como a apresentada neste caso clínico, é fundamental, de modo a que o médico esteja sensibilizado para formas de apresentação menos típicas, permitindo que o diagnóstico possa ser feito o mais precocemente possível.
Palavras-chave: Neuropatia amiloide familiar; Parestesia.
ABSTRACT
Introduction: Familial amyloid polyneuropathy (FAP) is a hereditary amyloidosis that presents with a progressive, mixed neuropathy (sensory, autonomic and motor). This case shows the importance of the family physician in the diagnosis of this pathology, while highlighting the main difficulties of performing the diagnosis at advanced ages, and when there are no other documented cases in the family.
Case report: A 71-year-old woman with a personal history of ductal carcinoma in situ, carpal tunnel syndrome, and adenomatous multinodular goiter. Over the past 5 years, she has had multiple non-specific complaints, including chronic constipation, neuralgia and paresthesias of the hands and feet, progressive loss of control of the urinary sphincter, progressive locomotor activity deterioration, and weight loss. There was no family history of PAF except a 65-year-old sibling diagnosed with late-onset PAF with complaints of paresthesias of the hands and feet. This information alerted to the hypothesis of a diagnosis of FAP, which would justify the multiple complaints of the patient, and which was confirmed by genetic testing.
Comment: This clinical case describes a series of particularities that should alert us to the fact that this pathology may arise with a different pattern than the one described by Corino de Andrade: the age of onset, the absence of affected parents, as well as the presence of the disease in families that are not originally from Póvoa de Varzim. Thus, the knowledge of late variants of PAF as the one presented in this case report is fundamental to raise awareness on less typical clinical presentations, thus allowing an early diagnosis.
Keywords: Familial amyloid neuropathies; Paresthesia.
Introdução
A polineuropatia amiloide familiar (PAF) é uma amiloidose hereditária autossómica dominante, que resulta de uma mutação no gene da transtirretina (TTR). Esta patologia manifesta-se geralmente na idade adulta através de uma neuropatia mista (sensitiva, autonómica e motora), com atingimento multiorgânico, de caráter progressivo.1
Esta patologia foi descrita pela primeira vez em 1952, por Corino de Andrade, a partir da observação de doentes oriundos do litoral norte, na região da Póvoa do Varzim. Contudo, tem-se verificado que a idade de início da doença e a apresentação inicial é mais variável que as descrições iniciais de Corino de Andrade.2
O caso clínico relata a importância e o lugar privilegiado do médico de família para o diagnóstico desta patologia, enquanto realça as principais dificuldades nesse diagnóstico em idades mais tardias, bem como quando não existe história familiar conhecida.
Descrição do caso
Identificação e caracterização do estado de saúde
Mulher de 71 anos, de raça caucasiana e reformada, que trabalhou durante muitos anos como operária numa fábrica têxtil. Nasceu e viveu toda a sua vida em Campo-Valongo, no Porto, no seio de uma fratria de sete irmãos. Está casada há 48 anos, união na qual teve dois filhos e uma filha.
Esta utente é frequentadora de consultas na USF São João de Sobrado, sendo também seguida em consultas hospitalares, pelos seus antecedentes pessoais de:
Carcinoma ductal in situ, diagnosticado em 2010. Fez tumorectomia e seguimento durante os seis anos seguintes, sem recidiva locorregional, mantendo desde então vigilância clínica e imagiológica anual.
Síndroma do túnel cárpico (STC) bilateral, diagnosticado em 2013, com confirmação por eletromiografia que mostrou STC grave à esquerda e muito grave à direita. Fez cirurgia à direita em 2013; manteve, no entanto, as queixas de parestesias da mão.
Bócio multinodular adenomatoso, diagnosticado em 2015. Fez tiroidectomia total por não ter sido possível excluir carcinoma papilar após punção biópsia aspirativa. Sob suplementação com levotiroxina e vigilância clínica e imagiológica anual.
Fratura bimaleolar do pé esquerdo após queda acidental com trauma, em janeiro de 2016. Fez redução e osteossíntese com placa e parafusos e retomou a marcha com canadiana. Fez tratamentos de medicina física e reabilitação e recuperou marcha autónoma, sem auxiliares da marcha, ainda que mantendo alguma instabilidade da marcha.
Tem o Programa Nacional de Vacinação atualizado, sem hábitos tabágicos ou alcoólicos. Na sua história ginecológica e obstétrica refere-se menarca aos 13 anos e menopausa aos 40 anos, com quatro gravidezes, três partos e um aborto espontâneo (1º trimestre, de causa desconhecida).
História da doença atual
Concomitantemente às patologias crónicas descritas, esta utente tem recorrido a consultas na sua unidade de saúde por apresentar diferentes sintomas inespecíficos, os quais foram avaliados e orientados de acordo com o seu aparecimento. Em 2013 apresentou, pela primeira vez, queixas de parestesias do primeiro e segundo dedos da mão direita, sendo na altura encaminhada para consulta de ortopedia por queixas compatíveis com STC. Além de a cirurgia não ter permitido uma melhoria significativa destas queixas, três anos depois a utente apresenta queixas semelhantes na mão esquerda e em ambos os pés e alterações da força ou da sensibilidade para o calor ou frio, com deterioração significativa da capacidade de locomoção (com história de queda acidental e trauma do pé esquerdo em janeiro de 2016). Desde 2014 que apresenta alteração do trânsito intestinal, começou por manifestar alternância entre dejeções diarreicas e obstipação e, posteriormente, manteve um perfil obstipante (oito em oito dias), padrão ao qual se adaptou no seu quotidiano, recorrendo ao seu médico assistente apenas quando apresentava episódios de dejeções diarreicas exuberantes, compatíveis com quadros de gastroenterite. Em 2015 recorreu à sua médica de família por queixas de incontinência urinária de esforço; na altura recusou ser encaminhada para consulta de uroginecologia e iniciou tratamento com anticolinérgico (cloreto de tróspio), apenas com melhoria parcial das queixas.
Em fevereiro de 2016, a utente procura a sua médica de família com o receio de poder ter PAF, uma vez que foi feito este diagnóstico a um dos seus irmãos. Este irmão residente na Suíça, informou do seu diagnóstico recente e deu indicação aos irmãos para realizarem o teste genético para esta patologia.
Nesta data, a utente apresentava-se com um bom estado geral, normotensa, sem alterações à auscultação cardíaca ou pulmonar e com um peso de 71Kg (tendo perdido 15Kg nos últimos 12 meses, apesar de apetite preservado). Ao exame neurológico apresenta-se vígil, colaborante e orientada no espaço e tempo, com hipostesia álgica em ambas as mãos e pés (principalmente nos hálux e tornozelos), ausência de sensibilidade vibratória em ambos os pés e mãos, sem défices motores (apesar de difícil avaliação pós-trauma do pé esquerdo), reflexos aquilianos abolidos e rotulianos fracos. Capaz de marcha sem apoio, mas com grande dificuldade.
Perante a sintomatologia apresentada ao longo dos anos e a sua evolução surge a hipótese de diagnóstico de PAF, capaz de abranger as múltiplas queixas e que não tinha sido ainda equacionada. Neste sentido, a utente foi orientada para a consulta de neurologia/paramiloidose do Centro Hospitalar do Porto. A primeira avaliação clínica na consulta foi sugestiva que a utente poderia ser portadora da doença. Nesse sentido foi pedido o teste genético para TTR Met 30, o qual teve um resultado positivo, confirmando o diagnóstico de PAF com início tardio. Posteriormente, a utente fez biópsia da glândula salivar para confirmar a presença de substância amiloide, cujo resultado foi positivo.
Caracterização familiar
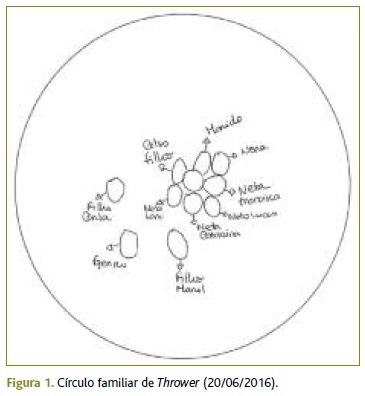
No âmbito da avaliação familiar, esta utente pertence a uma família nuclear, no estádio VIII do ciclo de vida de Duvall; vive numa moradia própria com o marido e com o filho mais velho, que retornou a casa dos pais após viuvez. Através do Apgar Familiar de Smilknstein pretendeu-se avaliar a forma como a doente sentia a sua posição na família naquele momento, sendo que em todas as questões a doente respondeu «algumas vezes», classificando a sua família com moderada disfunção. Através do círculo familiar de Thrower (Figura 1) constata-se uma boa relação de proximidade com o marido e com o filho mais velho com quem vive, superior em relação aos restantes; são estes os dois elementos da família a quem recorreria em primeiro lugar se necessitasse de algum apoio. É de destacar que a doente não representou, neste círculo, nenhum dos seus irmãos.
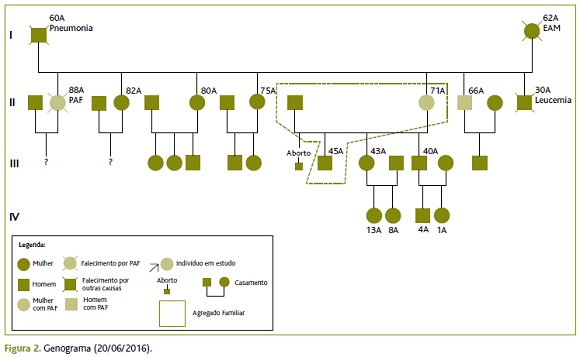
Em relação à sua história familiar (genograma - Figura 2), a utente refere que tanto entre os seus avós como nos pais nunca houve sintomatologia deste tipo, sendo que o pai faleceu aos 60 anos por patologia respiratória (trabalhou como pedreiro) e a mãe faleceu aos 62 anos por patologia cardíaca. O seu irmão, que vive na Suíça, começou com queixas de parestesias das mãos e pés aos 65 anos e no ano seguinte foi-lhe diagnosticada PAF. A irmã mais velha foi diagnosticada aos 88 anos com a mesma doença com recurso a teste genético (também no seguimento do diagnóstico deste irmão), apesar de ter sido avaliada previamente em consultas hospitalares, durante seis anos, por queixas de alterações da sensibilidade dos membros, sempre sem nenhum diagnóstico definitivo.
Comentário
A PAF, «uma forma peculiar de neuropatia», foi caracterizada pelo Professor Corino de Andrade como uma doença insidiosa, com início na 3ª ou 4ª décadas de vida, a que se segue a morte num período de sete a dez anos2 (10 a 20 anos, atualmente),3 sendo a PAF tipo I (associada à mutação Val30Met) a mais comum no Oeste Europeu e no Japão.4 Hoje sabe-se que é uma patologia com caráter hereditário (autossómica dominante) relacionada com a deposição sistémica de fibras de amiloide no tecido conjuntivo, provocada pela mutação de uma proteína em que a valina é substituída pela metionina na posição 30 no cromossoma 18.1
A evolução natural da doença caracteriza-se por alterações sensitivas, motoras e autonómicas. As alterações sensitivas começam quase invariavelmente nos pés, com progressão ascendente e comprometendo posteriormente as mãos, inicialmente de forma centrípeta, sendo que numa fase mais tardia da doença evolui geralmente para um padrão em «meia» e em «luva». O compromisso motor é mais tardio, causando alterações da massa muscular e diminuição da força. A atrofia dos músculos das mãos pode conferir uma configuração em forma de garra. Em alguns casos, a primeira manifestação da doença pode ser a síndroma de túnel cárpico.3,5 Outras das manifestações mais frequentes são a perda ponderal, consequência da deficiente absorção intestinal.6
A disfunção autonómica presente nos doentes com PAF inclui disfunção sexual e disfunção esfincteriana, que se caracteriza por necessidade de esforço no início da micção e interrupção do jato, progredindo para quadros de retenção urinária e, posteriormente, incontinência.7 A infiltração de amiloide no miocárdio leva a cardiomiopatia, devido a disfunção do sistema de condução e perturbações decorrentes da disautonomia, nomeadamente alterações da pressão arterial e da frequência cardíaca.8-10 As perturbações gastrointestinais caracterizam-se frequentemente pelo aparecimento de obstipação, seguida de períodos alternados de obstipação-diarreia e culminando com a diarreia crónica, podendo surgir em estadios avançados a incontinência fecal. As alterações renais são características de fases mais avançadas da doença, podendo progredir até insuficiência renal terminal com necessidade de hemodiálise. As manifestações oculares mais frequentes são queratoconjuntivite sicca, glaucoma, opacidades do vítreo e alterações na vasculatura da conjuntiva.3 Pode ocorrer atingimento do sistema nervoso central, cutâneo, pulmonar, entre outros órgãos e sistemas.
Devido à multiplicidade de sintomas existentes, o diagnóstico de PAF com base apenas na clínica é um desafio, sendo por isso fundamental a história familiar e os meios complementares de diagnóstico (patologia, neurofisiologia e genética).
O caso clínico descrito apresenta uma série de particularidades que nos devem alertar para o facto de esta patologia não ter apenas o padrão descrito por Corino de Andrade: esta família vive no concelho de Valongo, o que alerta para o facto de a PAF ser uma patologia em expansão por todo o país e não uma patologia característica da região da Póvoa de Varzim.
A idade de apresentação dos primeiros sintomas foi aos 69 anos, com um padrão sensitivo e disautonómico e, desde então, surgiram sintomas compatíveis com atingimento gastrointestinal, genitourinário e, mais recentemente, atingimento motor. Qualquer um dos sintomas apresentados poderia ser interpretado como alterações fisiológicas do envelhecimento ou como uma patologia orgânica degenerativa.
É também importante realçar que não existe história de progenitores afetados, o que nos faz refletir sobre questões de penetrância do gene mutado. De facto, os casos inicialmente descritos de PAF apresentavam uma ≠penetrância elevada, a doença era conhecida na família há muitas gerações, ocorriam manifestações de sintomas em todos os portadores e caracterizava-se por uma idade de início precoce (3ª ou 4ª década de vida). Contudo, têm sido descritos casos de baixa penetrância, que são casos aparentemente esporádicos, a mutação é silenciosa em muitos portadores, sendo que nestes a idade de início tem sido mais tardia, como se verificou no caso clínico apresentado.
Neste caso clínico a suspeita de PAF surgiu apenas após o diagnóstico do irmão, que aos 65 anos apresentou sintomas semelhantes e, após estudo genético, foi identificada a mutação TTR Met 30. Nessa altura, a família foi alertada para a importância da realização de estudo genético, pelo que, após integração deste quadro clínico aparentemente inespecífico, surgiu imediatamente a suspeita de que a utente poderia ser portadora da mesma mutação. Deve também ser realçado o caso de outra irmã, que estaria internada para estudo por um quadro avançado de astenia, perda de força e disautonomia e que, neste contexto, se percebeu que também seria portadora da doença.
Ainda no que diz respeito ao impacto familiar da PAF, é essencial destacar que a confirmação do diagnóstico tem implicações em relação à descendência da doente. Assim, os seus filhos têm indicação para realizar o estudo genético, possibilitando o diagnóstico precoce.
A existência de tratamentos modificadores da doença, como o transplante hepático e o mais recente tafamidis, exclusivamente para doentes em fases iniciais, tornam mais dramático o atraso do diagnóstico.
Assim, o conhecimento da existência de variantes tardias da PAF, como neste caso clínico, é fundamental para que os profissionais de saúde estejam alerta para formas de apresentação menos típicas, de modo a que o diagnóstico possa ser feito o mais precocemente possível.
A suspeita diagnóstica e a investigação dirigida são duas situações em que o médico de família tem um papel fundamental e privilegiado, tal é a sua relação de proximidade com o doente, a sua família e a comunidade. Devido ao curso negativo e fatal da PAF, é constante a necessidade de investigação nesta área ao nível de suporte sintomático com impacto na melhoria da qualidade de vida, bem como de meios que impeçam a progressão da doença.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol. 2005;62(7):1057-62. [ Links ]
2. Andrade C. A peculiar form of peripheral neuropathy: familial atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75(3):408-27. [ Links ]
3. Conceição I. Clínica e história natural da polineuropatia amiloidótica familiar [Clinical manifestations and natural history of familial amyloidotic polyneuropathy]. Sinapse. 2006;6(Suppl 1):86-91.
4. Koike H, Sobue G. Late-onset familial amyloid polyneuropathy in Japan. Amyloid. 2012;19 Suppl 1:55-7. [ Links ]
5. Munar-Qués M. Polineuropatía amiloidótica familiar 2003 [Familial amyloid polyneuropathy 2003]. Med Clin (Barc). 2003;121(3):100-1. Spanish
6. De Carvalho M, Conceição I, Bentes C, Luís ML. Long-term quantitative evaluation of liver transplantation in familial amyloid polyneuropathy (Portuguese V30M). Amyloid. 2002;9(2):126-33. [ Links ]
7. Andrade MJ. Alterações vesico-esfincterianas na polineuropatia amiloidótica familiar [Dissertation]. Porto: Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto; 2001. [ Links ]
8. Mathew V, Olson LJ, Gertz MA, Hayes DL. Symptomatic conduction system disease in cardiac amyloidosis. Am J Cardiol. 1997;80(11):1491-2. [ Links ]
9. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75-84. [ Links ]
10. Mörner S, Hellman U, Suhr OB, Kazzam E, Waldenström A. Amyloid heart disease mimicking hypertrophic cardiomyopathy. J Intern Med. 2005;258(3):225-30. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Andreia Sofia Ferreira Marques
E-mail: andreiasofiamarques@gmail.com
http://orcid.org/0000-0003-0491-3424
Conflito de interesses
As autoras declaram não possuir quaisquer conflitos de interesse.
Recebido em 02-12-2016
Aceite para publicação em 04-06-2018

