









 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Aortic dissection may occur in individuals with inhered disorders of connective tissue, including those caused by fibrillin mutations (Marfan syndrome), collagen mutations (Ehlers-Danlos syndrome), transforming growth factor ² receptor mutations (Loeys-Dietz syndrome) and in those in which a connective tissue disorder is postulated but not proven, such as bicuspid aortic valve syndrome and Turner syndrome (TS).
TS, which has a prevalence at birth of 1/2000 girls, is due to complete or partial absence of an X chromosome and is almost invariably associated with growth retardation and ovarian failure with infertility.(1)
The most serious life-threatening issues of X-chromosome haploinsufficiency are caused by lesions of the cardiovascular system. Congenital cardiac abnormalities are described in up to 50% of cases and cardiovascular complications are those which are mostly responsible for early excess mortality.(2) Life expectancy may be reduced by more than 10 years.(2) The incidence of these congenital abnormalities is higher in 45,X monosomies and rarer (20%) in mosaics or in people with a structural X chromosome abnormality.(3)
Cardiovascular findings occurring in TS might be congenital or acquired.(4) Congenital cardiovascular defects are associated mainly with left-sided malformations. Acquired cardiovascular problems in women with TS may be caused by a number of risk factors, including central obesity, impaired glucose tolerance, insulin resistance and hyperlipidemia.(4,5)
In TS patients, dissections are usually acute, associated with chest, neck and/or back pain and often involve proximal rupture, hemopericardium and tamponade.(6)
We describe a case of type A aortic dissection occurring in a woman with TS, highlighting the need to prioritize investigation in those patients to avoid a potentially catastrophic aortic scenario.
Case report
A 48-year-old woman was diagnosed with TS (45,X) at the age of 20 years after an evaluation for short stature and delay of pubertal development. She was on sex hormone replacement therapy, since her TS diagnose and had no other relevant medical history, besides her genetic disease. The patient was admitted to our institution with an intense and acute thoracic pain. On admission, the patient was found to be with hypertension (180/90 mmHg) and normocadiac rhythm. She was overweight (BMI 27 kg/m2), with no prior history of pregnancy. The patient’s most recent echocardiogram six months prior to the dissection, showed no abnormalities.
There were no signs of acute cardiac ischemia on the electrocardiogram.
After the initial analytic study, an echocardiogram and a computed tomography angiography (CTA) were performed and demonstrated an acute Stanford type A dissection extending from midway to the left common carotid artery and the left subclavian artery to the distal aortic arch.
She was immediately taken to the operating room and was submitted to an emergency open repair - replacement of aortic arch (with reimplantation of innominate trunk and left carotid artery) using a 20-mm Dacron graft. The post-operative period was complicated by a surgical infection of the sternotomy - inflammatory signs of the incision, with leukocytosis. There was no agent isolated and the patient was treated with a cycle of gentamicin and vancomycin being discharged 10 days after.
She was medicated with an angiotensin receptor blocker, a diuretic and a ²-blocker to control the blood pressure.
The patient was admitted 3 months later with a chronic type B dissection and symptoms of mesenteric malperfusion - she complained of abdominal pain resistant to analgesic medication and persistent hypertension. The AngioTC showed the dynamic compression of true lumen and revealed that superior mesenteric artery arised from both false lumen and true lumen and left renal artery arised from false lumen. Therefore, she was submitted to a Thoracic EndoVascular Aortic Repair (TEVAR) (Medtronic Valiant 24*104 mm - Medtronic Vascular, Santa Rosa, CA) with a proximal landing of the stentgraft in zone 2 (Ishimaru classification); a vascular plug was also placed at the origin of the subclavian artery, and a covered stent was implanted in the left renal artery. She was discharged one week later with no symptoms. During follow-up, the 12 months CTA reveled a new tear at the distal portion of the stentgraft with a significant insinuation of this one into the false lumen and a proximal progression of the later (figure 1).
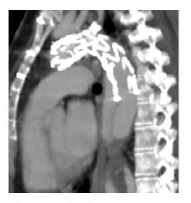
Figure 1 CTA reconstruction at 1year of FU showed a new tear at the distal portion of the stentgraft with an insinuation of it into the false lumen
Subsequently, we decided to perform a relining of the previous graft with a new stentgraft (Medtronic Valiant 26*100 mm - Medtronic Vascular, Santa Rosa, CA) - Figure 2.
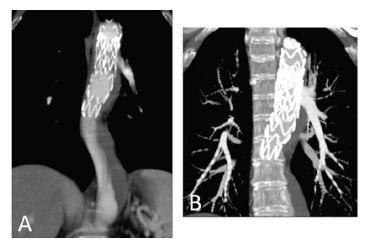
To improve our result, we blocked the retrograde perfusion of the false lumen ballooning the medium region of the graft in order to expand the graft into the false lumen - modified “knickerbocker technique”.(7) Final angiography showed a minimal distal false lumen reperfusion, which was completely occluded in the thoracic segment. The CTA performed one month after confirmed adequate relining of the graft and complete thrombosis of the false lumen in the thoracic aorta (figure 3). The last performed CTA was at 2 years follow-up and showed imagiological stability and thrombosis of false lumen at the thoracic aorta level.
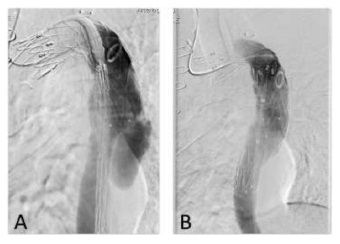
Figure 2 A -Angiographic images before the relining of the previous endograft with a Medtronic Valiant 26*100 mm thoracic distal component stent graft with a distal landing zone above the celiac artery; B - Final angiographic image
Discussion
Turner syndrome (TS) is a genetic disorder with a characteristic phenotype that occurs as a result of a structurally abnormal or absent X chromosome and is the only monosomy compatible with life.(1)
There are variable Turner karyotypes, such as monosomy X (45,X; the most prevalent karyotype), mosaic karyotype, isochromosome X, ring X chromosome or deletions. In all these cases, significant portions of the X chromosome are deleted.(8)
The diagnosis of TS is associated with key clinical features, including short stature and gonadal failure (the cardinal features) and congenital cardiovascular (CV) defects as well as an abnormal karyotype.(1)
Cytogenetic screening studies indicate that TS occurs in about 1 in 200 conceptions but approximately 90% do not survive delivery.(9) Most die in the antenatal period due to left ventricular outflow tract pathology. Major defects in cardiac and aortic development during fetal life are associated with miscarriage in most cases of fetuses with 45,X karyotype.(1) For the 10% who survive, congenital cardiac abnormalities are described in approximately 30%-50%.(10) Bicuspid aortic valve (BAV) is the commonest abnormality, reported in about 20%-30% of these patients.(11) Coarctation of the aorta (CoA) is present in about 4%-12%, often presenting with neonatal collapse in the first week of life or diagnosed later on in childhood due to hypertension or limb claudication.(12)
It is increasingly accepted that TS is associated with a higher risk of aortic dilatation and aortic dissection, which can be fatal if not recognized early.(13) In this context, the most serious consequences of X-chromosome haploinsufficiency involve the CV system. Indeed, CV defects, present in up to 50% of the TS population, are the major cause of premature death and contribute to a standardized mortality ratio three times higher than in the general female population. Up to 90% of aortic dissections have predisposing CV risk factors (bicuspid aortic valve (BAV), aortic dilatation and CoA) that can be identified with cardiovascular magnetic resonance (CMR) imaging. Therefore, imaging studies (both echocardiography and CMR) are crucial for the timely detection of subclinical CV disease and should preferably be ordered before the onset of symptomatic and irreversible organ damage.(14)
Although rare, aortic dissection is a major concern in TS. TS is considered one of the high-risk conditions that causes aortic dissection(15). It has an estimated incidence of 40 cases per 10000 patients, but is often fatal. It occurs at a much earlier age. In the general population, the peak incidence of dissection occurs between ages 50 and 80 and in TS population median age is 35, with higher incidence rates at ages 20-29 and 30-39.(6)
An epidemiological study of Danish and Swedish records(6) found the median age of onset of aortic dissection or rupture in TS to be 35 years and projected an annualized incidence of approximately 15 cases/100000 TS for individuals <20 years of age, 73-78 cases/ 100000 for women 20-40 years old and 50/100000 for older women with TS. This is against a background rate of 6/100000 in the general Danish population, with average age of 71 years and male:female ratio of 2:1.
The majority of dissections in TS occur in the ascending aorta.(13) Choong Wong et al, in there review of 122 published cases, conclude that 66 cases (54%) experienced dissection in the ascending aorta, 20 (16.4%) in descending aorta, 10 (8.2%) in ascending and descending aorta and the remainder at other sites (e.g., common carotid, abdominal in combination with either ascending or descending aorta) or sites not reported.
A major risk factor for aortic dissection in TS is aortic dilatation. Other possible risk factors include age, karyotype and other cardiovascular abnormalities, hypertension and pregnancy.(16)
The last two are correlated with a higher risk of dissection due to an association with increased aortic root diameter and to physiological adaptation, which imposes a higher cardiac workload. However, up to 25% of cases have no apparent risk factors other than TS itself, suggesting that TS phenotype may also include a primary aortopathy.(5,6)
According to Hjerrild et al. study,(11) the report of ambulatory 24-h blood pressure monitoring in TS revealed approximately 60% of adults with TS to be hypertensive. Although, in a literature survey of all published cases in 2007, only 50% of TS patients with dissection were hypertensive.(17) As some of those patients may have received anti-hypertensive treatment, the contributing role of hypertension in dissection is unknown.(17)
In recent years, increasing numbers of women with TS have become pregnant via assisted reproduction. Pregnancy is a high-risk period for aortic dissection in TS.(18) The estimated risk of death from aortic dissection during that period is 2%.(19) Due to that fact, aortic dissection risk evaluation should now be mandatory for all patients with TS prior to consideration of pregnancy.
As mentioned before, aortic diameter is a major risk factor for aortic dissection in TS and it is the principal risk stratification tool for that complication.(15,20)
An increased rate of aortic dissection in TS was initially attributed to the presence of CoA, but histological evidence of cystic medial necrosis in aortic tissue taken from patients with BAV, Marfan syndrome and TS indicates a shared etiology despite genetically diverse backgrounds.(17)
A study suggested that aortic disease in TS was similar to that in Marfan syndrome, characterized by ‘cystic medial degeneration’, which is the common pathophysiology for aortic root dilation.(17,21,22) However, there is no evidence to suggest that patients with TS experience progressive dilatation of the aorta over time, in the absence of predisposing risk factors, as seen in Marfan syndrome. It remains unknown whether the dilatation of the ascending aorta predicts aortic dissection in TS as it does in Marfan syndrome, and there are no current guidelines on what specific aortic diameter measurement should provoke concern in TS.(23)
Cardiovascular data of 233 patients (children and adults) with TS was analysed by Donadille and their colleagues(24); they concluded that at least one aortic diameter exceeded 32 mm in 28 patients (12%), but after indexation to body size, 91 (39%) patients from the cohort had at least one aortic diameter over 20 mm/m2. So, despite a normal ascending aortic diameter in absolute terms, after indexation to body size, patients with TS may have a dilated aorta.
The risk for aortic dissection in a TS patient with “normal” aortic dimension with no congenital heart disease, valvular or vascular abnormalities, as the previous case reported before is unknown. It is possible that the risk of aortic dissection for those TS patients are lower but it is still considered that these patients are at higher risk of aortic dissection and follow-up is essential. As so, complete cardiac evaluation has been recommended for all patients when TS is diagnosed.(3) Echocardiography has become the most common initial imaging test in the evaluation of cardiovascular disease and plays an important role in the diagnosis and follow-up of aortic diseases.(24) Still, CMR allows better visualization of thoracic aorta. The advantages of CMR include the ability not only to identify anatomic variants and aortopathies but also to assess branch artery involvement, and diagnose aortic valve pathology, as well as left ventricular dysfunction without exposing the patient to either radiation or iodinated contrast.(15)
Current medical treatment recommendations for patients with thoracic aortic disease implies strict control of hypertension, but also optimization of lipid profile, smoking cessation and control of the remaining cardiovascular risk factors.(15) Additionally, several observational, retrospective and interventional studies have supported the hypothesis that sex hormone replacement therapy exerts important beneficial effects on heart disease, diabetes and obesity in TS patients.(4)
Little evidence is available concerning the more adequate surgical treatment for type B dissections in TS patients being the best treatment modality for these patients not yet been established. Endovascular approach is generally recognized as an alternative to open surgery. The main goal is to seal the entry site, which induces thrombus and obliteration of the false lumen. Secondarily, the stent-graft provides compression of the false lumen.(26) In this case the stent-graft covered the entry site, but not all re-entry sites. There is no consensus about the length of the stent-graft. Some authors state that only coverage of the entry site is sufficient and a long stent-graft might increase the risk of spinal cord ischemia. This patient had previously a thoracic surgery and was treated by vascular surgery in an acute setting so the endovascular treatment was the less invasive procedure to obtain the success.
Though, endovascular treatment might be associated with postoperative adverse events, such as endoleak or stent graft migration, which demand a regular follow-up that could be troublesome for patients, especially young patients.
Conclusion
Despite the high prevalence of cardiovascular pathology affecting young women with TS, management of this disorder is still a challenge.
The etiology of aortic lesions remains poorly understood. Careful cardiovascular screening and follow-up using techniques such as echocardiogram and CMR are crucial, but usually this issue is still not adequately addressed in clinical practice.
As previously mentioned, risk for acute aortic dissection is highest in the third and forth decades in women with TS. Physicians must be aware of this particular risk in the group of patients suffering from this genetic disorder. Due to the lack of a consensus about the monitoring of these patients, they should be followed not only during childhood and adolescence, but also during adulthood to avoid potentially undetected lethal cardiovascular complications. Prospective studies are needed in order to establish the exact risk of aortic dissection, which imaging modalities to use, to identify patients with an elevated risk, and, if possible, to introduce procedures and/or medicine to lower the risk, both during pregnancy and normal life.
In this particular case, the authors are performing an annual clinical and imagiological (CTA) evaluation, as the patients has now 2 years of follow-up since the last intervention.