










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkJornal Português de Gastrenterologia
versão impressa ISSN 0872-8178
J Port Gastrenterol. vol.19 no.5 Lisboa set. 2012
Doença hepática autoimune na criança e no adolescente - dificuldades no diagnóstico
Autoimmune liver disease in children and adolescents - difficulties on diagnosis
Marta Riosa,∗, Teresa São Simãoa, José Ramón Vizcaínob, Margarida Medinaa, Fernando Pereiraa e Ermelinda Santos Silvaa
a Unidade de Gastrenterologia Pediátrica, Centro Hospitalar do Porto, Porto, Portugal
b Serviço de Anatomia Patológica, Centro Hospitalar do Porto, Porto, Portugal
*Autor para correspondência
Resumo
Introdução: A doença hepática autoimune (DHAI) pode envolver predominantemente os hepatócitos ou o epitélio dos ductos biliares intra/extra-hepáticos, ou ambas as estruturas (simultaneamente ou de forma sequencial), denominando-se, respetivamente, hepatite autoimune (HAI), colangite esclerosante primária (CEP) e síndrome de overlap (SO).
Objetivo: Conhecer as dificuldades de diagnóstico de DHAI numa população pediátrica.
Métodos: Estudo retrospetivo dos casos seguidos num hospital pediátrico nos últimos 19 anos.
Resultados: Foram incluídos 20 doentes (10 M, 10F): HAI-10, CEP-7, SO-3. A mediana de idades à data de aparecimento dos sintomas foi de 9,0 anos e à data de diagnóstico de 11,5 anos. Dos 10 doentes com HAI, 3 tinham score pré-tratamento de diagnóstico provável e todos score póstratamento de diagnóstico definitivo. Em 1 doente a presença de ds-DNA obrigou a diagnóstico diferencial com lúpus. Dos 7 doentes com CEP, apenas 2 apresentavam anomalias imagiológicas das vias biliares e 4 lesões histológicas ductulares. Três doentes apresentaram critérios de diagnóstico de SO, um de aparecimento sequencial.
Conclusões: As maiores dificuldades foram encontradas nos doentes com CEP em estadio inicial e nos doentes com SO, sobretudo no caso de apresentação sequencial. Destacam-se também as dificuldades em efetuar o diagnóstico diferencial entre HAI como entidade independente e outras doenças autoimunes multissistémicas com atingimento hepático, como o lúpus eritematoso sistémico.
Palavras-chave Hepatite autoimune; Colangite esclerosante primária; Síndrome de overlap; Criança
Abstract
Introduction: Autoimmune liver disease (AILD) may involve predominantly the hepatocytes or the intra/extra-hepatic bile duct epithelium, or both structures, naming respectively, autoimmune hepatitis (AIH), primary sclerosing cholangitis (PSC), and overlap syndrome (OS).
Aim: To identify the diagnostic difficulties of AILD in a paediatric population.
Methods: Retrospective study of the cases followed in a pediatric hospital in the last 19 years.
Results: Included 20 patients (10 M, 10F): AIH-10, PSC-7, OS-3. Median age at the time of onset of symptoms was 9,0 years and at the date of diagnosis was 11,5 years. Of the 10 patients with AIH, 3 had a pre-treatment score of probable diagnosis, and all had a post-treatment score of definitive diagnosis. In 1 patient the presence of dsDNA forced the differential diagnosis with lupus. Of the 7 patients with PSC, only 2 had abnormalities of the biliary tract on imaging, and 4 histological lesions in the ducts. Three patients had diagnostic criteria for OS, 1 with sequencial presentation.
Conclusions: The major diagnostic difficulties were encountered in patients with PSC in early stage, and in cases of OS, especially the one of sequential presentation. Also noteworthy are the difficulties of the differential diagnosis between HAI as an independent entity and other multisystem autoimmune diseases with liver involvement, such as systemic lupus erythematosus.
Keywords Autoimmune hepatitis; Primary sclerosing cholangitis; Overlap syndrome; Child
Introdução
A doença hepática autoimune (DHAI) é responsável por 2 a 5% dos casos de hepatopatia crónica na criança1,2. Resulta de intolerância imunológica contra as células hepáticas, com evolução para inflamação crónica e destruição progressiva3. Este processo pode envolver predominantemente os hepatócitos ou o epitélio dos ductos biliares intra e/ou extra-hepáticos, denominando-se respetivamente hepatite autoimune (HAI) e colangite esclerosante primária (CEP)3-5; a cirrose biliar primária (CBP) nunca foi diagnosticada em idade pediátrica. Aspetos sugestivos de ambas as doenças (HAI e CEP) podem estar presentes no mesmo doente, simultaneamente na altura do diagnóstico, ou em alturas diferentes ao longo da evolução da sua doença, designando-se nesse caso «síndrome de overlap» (SO)5-8. Na idade pediátrica é particularmente frequente o overlap HAI-CEP.
Existem scores de diagnóstico de HAI validados para adultos e que são aplicados na população pediátrica1,9-11, mas não existem critérios de diagnóstico bem definidos de CEP1,4,6,7. Para além das dificuldades por vezes notadas em afirmar o diagnóstico, a distinção entre HAI, CEP e SO pode ser muito difícil.
Na idade pediátrica há alguns estudos sobre HAI1,2,4,8,12-29, mas são mais raras as publicações sobre CEP e SO8,28-33.
O objetivo deste trabalho é conhecer as dificuldades de diagnóstico de DHAI numa população pediátrica.
Material e métodos
Estudo retrospetivo baseado na consulta dos processos clínicos das crianças e adolescentes com DHAI seguidos nas consultas de Hepatologia e de Gastrenterologia de um hospital pediátrico, no período de janeiro de 1992 a 31 de dezembro de 2010.
Parâmetros analisados: sexo, idade, aspetos clínicos, estudos laboratoriais, imagiológicos e histológicos e resposta ao tratamento.
O diagnóstico de HAI foi estabelecido segundo os critérios do Grupo Internacional para Estudo da HAI (IAIHG), publicados em 19939 e revistos em 199910 e em 200811.
Em todos os doentes foi efetuado o seguinte estudo analítico: hemograma, bilirrubinas e enzimas hepáticas, proteinograma, imunoglobulinas e auto-anticorpos (auto-
Acs), estudo da coagulação, serologias víricas (anti-HAV, AgHbs, anti-HCV, EBV, CMV), _-1-antitripsina e ceruloplasmina séricas e doseamento do cobre urinário.
Foi efetuada biópsia hepática em todos, sendo o exame histológico realizado pelo mesmo anatomopatologista, à exceção dos casos 11 e 20 (biópsia realizada noutra instituição hospitalar). Após o diagnóstico, todos os doentes com HAI e SO efetuaram tratamento com imunossupressores (prednisolona 2 mg/kg/dia, máximo 60 mg/dia, e/ou azatioprina 0,5-2,5 mg/kg/dia) e todos com CEP e SO foram medicados com ácido ursodesoxicólico (AUDC) 15- 20 mg/kg/dia, em 2 tomas.
Resultados
Durante este período foram diagnosticados 20 casos de DHAI: 10 - HAI, 7 - CEP e 3 - SO. Cinco destes casos foram previamente publicados (casos 2, 6, 7, 8 e 18)13,31.
Sexo, idade e aspetos clínicos
Dez crianças/adolescentes eram do sexo feminino (50%).
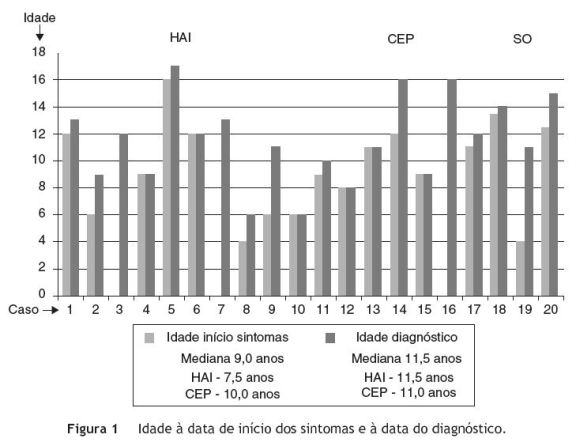
A mediana de idades à data de aparecimento dos sintomas foi de 9,0 anos (mínimo 4,0 e máximo 16,0) e à data do diagnóstico de 11,5 anos (mínimo 6,0 e máximo 17,0), ou seja, cerca de 2,5 anos depois do início da sintomatologia - figura 1. Verificou-se que no grupo de doentes com HAI, a mediana de idades à data de aparecimento dos sintomas foi inferior (7,5 anos) em relação aos doentes com CEP (10,0 anos) e que o diagnóstico foi efetuado mais tarde (mediana de idades: HAI - 11,5 anos e CEP - 11,0 anos) - figura 1.
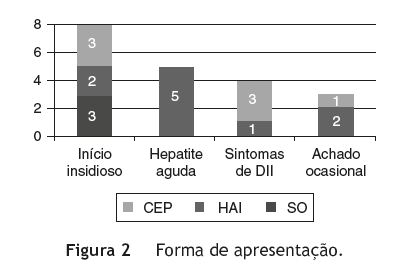
A forma de apresentação clínica foi variável - figura 2. No grupo de crianças/adolescentes com HAI, a doença manifestou-se sob a forma de hepatite aguda em 5, e teve início insidioso em 2 (tempo de evolução pré-diagnóstico de 3 anos no caso 2, e de 15 meses no caso 5). Em 2 doentes o diagnóstico foi efetuado após deteção acidental de elevação das transaminases em análises efetuadas por outros motivos: hematúria microscópica (caso 3) e pielonefrite aguda (caso 7). No grupo de crianças/adolescentes com CEP, o diagnóstico foi efetuado simultaneamente com o diagnóstico de colite ulcerosa (CU) em 3 doentes (casos 11, 13, 15) e, em outros 3, a doença teve início insidioso: dor abdominal intermitente e anorexia num doente com CU já conhecida (caso 12); dor abdominal esporádica e, 4 anos depois, aparecimento de icterícia, prurido e colúria (caso 14); icterícia, prurido, astenia e anorexia com um ano de evolução (caso 17). Num doente deste grupo o diagnóstico foi efetuado após deteção de elevação das transaminases em análises realizadas para estudo de obesidade (caso 16). No grupo de doentes com SO, em todos a doença teve início insidioso: no primeiro caso manifestou-se inicialmente por amenorreia secundária, no segundo caso por prurido e no terceiro caso por icterícia.
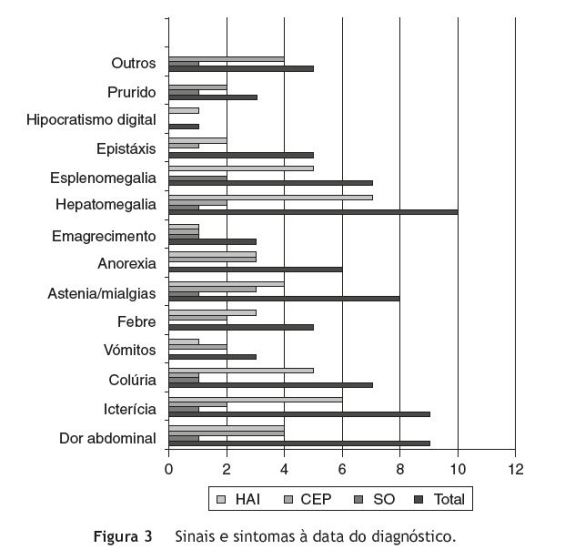
Os sinais/sintomas mais frequentes foram hepatomegalia, dor abdominal, icterícia e astenia/mialgias não havendo diferenças entre os 3 subgrupos de doenças, à exceção do prurido que só se verificou nos casos de CEP e/ou SO - figura 3.
Hepatite autoimune (Casos 1 a 10 - tabela 1)
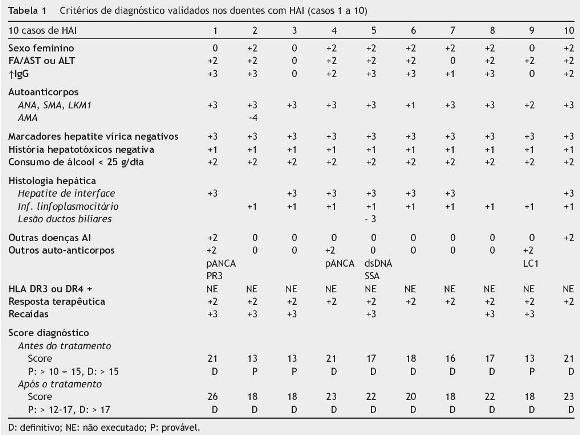
No grupo de 10 crianças/adolescentes com HAI, registaram-se 3 doentes do sexo masculino, não cumprindo o primeiro critério do score de diagnóstico10.
Em 2 crianças/adolescentes foi detetado outro tipo de doença de etiologia autoimune (AI): diagnóstico simultâneo de CU (caso 1) e diagnóstico prévio de trombocitopenia AI (caso 10).
Em 2 doentes, a relação fosfatase alcalina (FA)/transaminases foi superior a 3. Em 2 casos, não se verificou hipergamaglobulinemia. Em todos os doentes com HAI detetou-se, pelo menos, um dos auto-Acs habitualmente associados a esta patologia: ac anti-nuclear (ANA) - 8 (80%); ac anti-músculo liso (SMA) - 7 (70%), ac anti-microssoma hepático e renal (anti-LKM1) - 1 (10%). Os títulos variaram entre 1/40 e 1/1280. O caso 9 apresentava inicialmente títulos negativos que entretanto positivaram numa segunda amostra. Uma doente tinha ac antimitocondrial (AMA) positivo (caso 2), o que correspondeu a uma pontuação negativa no score de diagnóstico de HAI10. Uma doente com ANAs positivos, apresentava também dsDNA e SSA positivos (caso 5). Foram detetados outros tipos de auto-acs: ac anti-citoplasma dos neutrófilos (ANCA) em 2 doentes (20%), anti-citosol hepático tipo 1 (anti-LC1) em um doente (10%) e anti-proteinase 3 num doente (10%).
Em todas as crianças/adolescentes foram excluídas outras causas de doença hepática crónica.
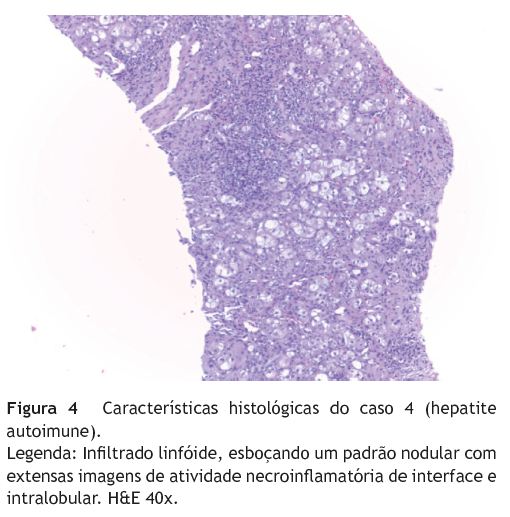
Verificou-se que todos os doentes apresentavam, pelo menos, uma das características histológicas típicas da HAI: hepatite de interface em 7 e infiltrado linfoplasmocitário em 9 - figura 4. Um doente (caso 5) apresentava também lesão dos ductos biliares.
Observou-se uma boa resposta ao tratamento imunossupressor em todos os doentes, tendo-se verificado recaída após a sua redução ou suspensão em 6 casos.
O somatório da pontuação de cada critério de diagnóstico correspondeu a um score de diagnóstico definitivo em 7 doentes e provável em 3. Após avaliação da resposta terapêutica aos imunossupressores o score de diagnóstico tornou-se definitivo em todos os doentes.
Colangiopatia esclerosante primária (Casos 11 a 17 - tabela 2)
Em relação aos 7 doentes com CEP, verificou-se que um era do sexo feminino. Quatro das 7 crianças/adolescentes tinham CU associada (em 3 diagnosticada simultaneamente e numa diagnosticada 3 anos antes). Uma criança apresentava ainda fenómenos de vasculite.
Em todos, detetou-se aumento da GGT e/ou FA, pelo menos 3 vezes superior ao limite superior do normal. A IgG estava aumentada em 3 doentes. No grupo de doentes com CEP detetaram-se os seguintes auto-Acs: ANA - 3 (43%), SMA - 5 (71%), ANCA - 2 (29%) e anti-proteinase 3 - 1 (14%). Os valores das titulações variaram entre 1/40 e 1/320. Também neste grupo, num doente os ANA foram negativos no primeiro estudo analítico e só posteriormente positivaram (caso 15).
Em relação aos aspetos imagiológicos, observou-se ectasia da via biliar principal ou das vias biliares intra-hepáticas em 2 doentes. Foi realizada colangiografia em pelo menos 4 doentes (CPRE - 2, colangioRM - 2): num doente em que foi tentada a CPRE, não foi conseguida a canulação biliar, tendose complicado de pancreatite aguda (caso 14); dos restantes 3, apenas se verificaram sinais de colangite esclerosante num doente.
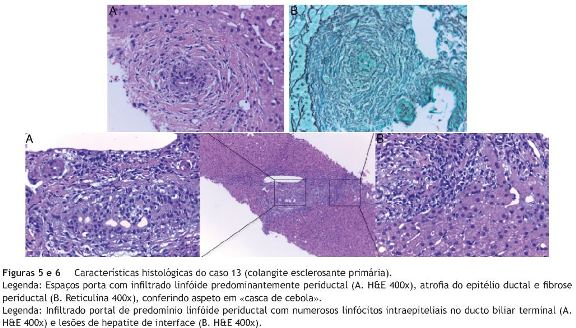
Relativamente aos aspetos histológicos, verificou-se alteração dos ductos biliares em 4 doentes (inflamação periductular nos 4 e proliferação ductular em 1) - figuras 5 e 6. Em 3 observou-se também inflamação dos espaços-porta e/ou hepatite de interface - figura 6. O relatório anatomopatológico de um dos doentes não estava disponível (caso 15).
Em todos os doentes com CEP verificou-se melhoria clínica e analítica após iniciarem tratamento com AUDC; em 4 foi associada prednisolona, com ausência de resposta em um.
Síndrome de overlap HAI-CEP (Casos 18 a 20 - tabela 3)
Caso 18 - Nesta doente do sexo feminino, previamente publicada30, a doença manifestou-se aos 13 anos por amenorreia secundária, e, 4 meses depois, por astenia, mialgias, perda ponderal, colúria e hepatoesplenomegalia. Os estudos complementares revelaram transaminases aumentadas, relação FA/transaminases < 1,5, hipergamaglobulinemia, ANA 1/2560 e, 2 anos depois, ANCA 1/1280 e evidência histológica de hepatite de interface e de proliferação de neoductos. Verificou-se resposta parcial à terapêutica imunossupressora e ao AUDC. O score pré e pós-tratamento foi concordante com diagnóstico definitivo de HAI. Porém, destacava-se elevação significativa da GGT, ausência de melhoria, após otimização da terapêutica imunossupressora e evidência ecográfica de ectasia das vias biliares intrahepáticas. Seis meses depois, efetuou CPRE que mostrou imagens sugestivas de colangite esclerosante, confirmando o diagnóstico concomitante de CEP e, por isso, de overlap HAICEP. Esta CPRE complicou-se de pancreatite e de colangites de repetição. Anos mais tarde e, já após ter sido submetida a transplante hepático, foi-lhe diagnosticada CU.
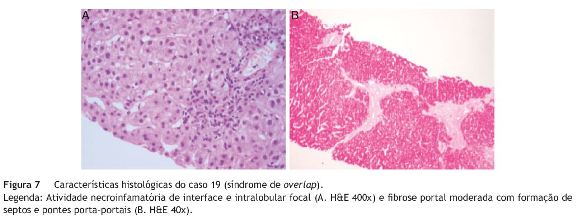
Caso 19 - Doente do sexo masculino, com uma avó paterna com CBP, que apresentava prurido desde o 1.◦ ano de vida (mais intenso a partir dos 4 anos), atribuído inicialmente a «alergias alimentares». Foi admitido na consulta de Hepatologia aos 11 anos de idade, e dos exames complementares efetuados nesta altura, destacava-se: bilirrubinas normais, elevação das enzimas hepáticas com predomínio da GGT e FA (AST 96 UI/L, ALT 165 UI/L, FA 577 UI/L, GGT 219 UI/L); ANA e SMA positivos; ecografia abdominal sem alterações; histologia hepática com evidência de fibrose nos espaços-porta, septos interportais, infiltrado portal misto com linfócitos e eosinófilos, esboço de fibrose periductal e neoformação ductular. Cumpriu terapêutica imunossupressora (prednisolona + azatioprina) durante 6 meses, sem melhoria significativa dos parâmetros analíticos, pelo que foi suspensa. Pela hipótese de se tratar de CEP, manteve-se apenas tratamento com AUDC. Aos 15 anos de idade verificou-se uma maior elevação das enzimas hepáticas e, um ano mais tarde, aparecimento de icterícia e agravamento do prurido, com o seguinte estudo analítico: BT 3,34 mg/dL, BD 2,96 mg/dL, AST 194 UI/L, ALT 543 UI/L, GGT 310 UI/L, hipergamaglobulinemia, ANA e SMA positivos e ANCA negativo. Nesta altura, a histologia hepática mostrava atividade necroinflamatória de interface e intralobular focal - figura 7. Por cumprir critérios de diagnóstico definitivo de HAI (score pré-tratamento 16) iniciou tratamento imunossupressor, desta vez com resposta francamente favorável. Este caso foi classificado como overlap HAI-CEP de apresentação sequencial (CEP seguida de HAI).
Caso 20 - Doente do sexo feminino que teve um primeiro episódio de icterícia colestática, sem prurido, aos 12 anos de idade (BT 10,2 mg/dL, BD 4,0 mg/dL, AST 117 UI/L, ALT 119 UI/L, GGT 185 UI/L). Nesta altura, foi confirmado o diagnóstico de síndrome de Gilbert por estudo molecular. A icterícia diminuiu, passando a ser apenas de bilirrubina livre (BT < 5 mg/dL) e as enzimas hepáticas normalizaram. Dois anos mais tarde teve novo episódio de icterícia colestática, com enzimas hepáticas elevadas e foi notada esplenomegalia, confirmada por ecografia abdominal, que mostrou também um fígado heterogéneo. Destacava-se a presença de trombocitopenia e ANA, SMA e Acs antitiroideus positivos, com IgG normal. O doseamento de _-1-antitripsina e a ceruloplasmina séricas foram normais, tal como o doseamento enzimático para as doenças de Gaucher e de Niemann-Pick tipo C. A histologia hepática revelou fibrose dos espaçosporta, hepatite de interface, atividade necroinflamatória lobular e intensa colestase hepatocanalicular. Foi tratada com prednisolona, sem melhoria significativa, pelo que foi suspensa. A CPRE mostrou vias biliares intra e extrahepáticas com morfologia normal, mas com alguma pobreza dos canais intra-hepáticos de 2.a e 3.a ordem. Iniciou tratamento com AUDC, registando-se normalização das enzimas hepáticas e da bilirrubina conjugada. Esta doente cumpria critérios de diagnóstico de HAI, mas não respondeu favoravelmente à prednisolona. Por outro lado, havia algumas alterações sugestivas de CEP (elevação da GGT, pobreza de canais intra-hepáticos de 2.a e 3.a ordem na CPRE) e houve resposta ao tratamento colerético. Apesar de atualmente ter enzimas hepáticas normais, foi associada azatioprina, por cumprir critérios de diagnóstico definitivo de HAI (score 17).
Discussão
Sexo, idade e aspetos clínicos
Embora a ocorrência de patologia AI predomine no sexo feminino, nesta série 10 (50%) das crianças/adolescentes eram do sexo masculino, a maior parte com CEP (6) e 1 com SO. O envolvimento das vias biliares ocorre sobretudo no sexo masculino5,6,34 (86% nesta amostra), ao contrário da HAI que é mais frequente no sexo feminino1,4,14 (70% nesta amostra).
Apesar de, na maior parte dos casos, a doença se manifestar na adolescência, os primeiros sintomas ocorreram em idade escolar em 7 doentes e, em idade pré-escolar, em 2.
Verificou-se que houve um intervalo relativamente grande (2,5 anos) entre a mediana de idades à data do início da sintomatologia e à data do diagnóstico. Esta diferença foi particularmente evidente em 4 doentes (casos 2, 9, 14 e 19) - figura 1. Nestes doentes (um deles residente em S. Tomé e Príncipe) houve um atraso na referenciação para centros terciários, pelo que se salienta a importância do conhecimento desta patologia e da orientação destes doentes para centros com experiência.
Os casos de HAI manifestaram-se como hepatite aguda em metade dos doentes, tal como noutros estudos1,2,4,8,14, sendo um aspeto facilitador do diagnóstico. Salienta-se o caráter indolente e insidioso de alguns casos de DHAI (8 no total da amostra), também verificado em outras séries1,13,19,27,28, sobretudo no grupo de doentes com CEP e SO (CEP-3, SO-3), o que pode atrasar a valorização dos sinais e sintomas e, consequentemente, o diagnóstico. Destacamse os 3 casos, cujo diagnóstico foi efetuado de forma acidental, e os 4 casos em que o diagnóstico foi efetuado na sequência do estudo de sintomas sugestivos de DII, patologia associada a este tipo de doença hepática, particularmente a CEP, como observado em 4 dos 7 casos de CEP (57%) e descrito em 80% dos casos na literatura6. É fundamental valorizar os antecedentes pessoais e familiares do doente, sobretudo no que diz respeito à ocorrência de outras doenças AIs, tais como DII, tiroidite AI, trombocitopenia AI e doença celíaca1,3,4,6,17,34. A percentagem relativamente baixa de outras doenças AI verificada na amostra estudada (8/20, 40%) deveu-se provavelmente ao facto de não ter sido efetuado doseamento de Acs antitiroideus e rastreio de doença celíaca em todos os doentes.
Estudo laboratorial
A anomalia bioquímica mais vezes associada a HAI é a elevação das transaminases (3 a 50 vezes superior ao normal)1,4,6,13, como observado em todos os casos. Em alguns doentes, pode ocorrer também elevação ligeira da FA4-6,29, como se observou nos casos 3 e 7. A relação entre o valor da FA e a AST ou ALT inferior a 1,5 é um dos critérios de diagnóstico de HAI10, mas que não se verificou nestes 2 doentes.
A elevação da FA e GGT é a anomalia mais consistente com o diagnóstico de CEP4-6,14,35, como se verificou em todos os casos de CEP. Numa fase precoce da doença, e sobretudo em idade pediátrica, o valor destas enzimas pode, contudo, estar normal30,34. As transaminases estão ligeiramente aumentadas na maioria dos casos (em 3 dos 7 casos de CEP desta amostra), mas podem atingir valores tão altos como 50 vezes superior ao normal6,30.
A IgG está aumentada em 60-80% dos casos de DHAI2,4,6. Apesar de esta alteração ser característica, os valores normais não excluem o diagnóstico1,2,4,6,14, como se observou em, pelo menos, 30% (6/20) da amostra estudada.
Uma outra característica da DHAI é a deteção de auto-Acs circulantes que reagem contra certas proteínas nucleares, citoplasmáticas e membranares1,4,6,14. Os mais importantes são os ANA, SMA e anti-LKM1. Há outro tipo de auto-Acs identificados mais recentemente, também importantes: anti-citosol hepático tipo 1 (anti-LC1), mais comum em doentes mais jovens e o único que parece relacionar-se com a atividade da doença; anti-citoplasma dos neutrófilos (ANCA); antigénio solúvel do fígado (SLA); anti-fígado e pâncreas (anti-LP)4. A titulação destes últimos Acs não foi efetuada nos doentes mais antigos da amostra estudada. A presença de AMA é típica da CBP1,4,5,35, mas pode ocorrer em até 5% dos casos de HAI1,36, como se observou no caso 2, correspondendo a 10% dos casos de HAI nesta série.
É raro haver crianças saudáveis com auto-Acs positivos. Qualquer valor/titulação superior a 1/20 para ANA e SMA e 1/10 para anti-LKM-1 neste grupo etário é clinicamente relevante.1,3,4,14. Na amostra estudada foram observados apenas valores superiores a 1/40.
Após pesquisa de todos os auto-Acs referidos, continua a haver cerca de 20-30% de doentes com DHAI sem auto-Acs detetáveis1,3,5,34 (1/20 - 5% nesta série - caso 17). A prevalência e características de DHAI seronegativa ainda não estão bem definidas3,4.
Embora a identificação destes auto-Acs seja um dado de extrema importância para o diagnóstico de DHAI, não são específicos da doença, não se relacionam com o grau de atividade da mesma (à exceção do anti-LC1) e os níveis podem variar ao longo da sua evolução1,2,13,36-38, como se verificou em 2 casos em que o doseamento de ANA era negativo num primeiro estudo, tendo sido positivo posteriormente.
Imagiologia
A ecografia abdominal pode revelar sinais sugestivos de cirrose e/ou de hipertensão portal, e dilatação dos ductos biliares intra ou extra-hepáticos nos casos de CEP4,35. Em até 50% dos casos não são detetadas quaisquer anomalias, o que acontece sobretudo numa fase precoce da doença4. No grupo estudado, verificou-se ectasia das vias biliares em apenas 3 doentes (2 com CEP e um com SO) - tabela 4.
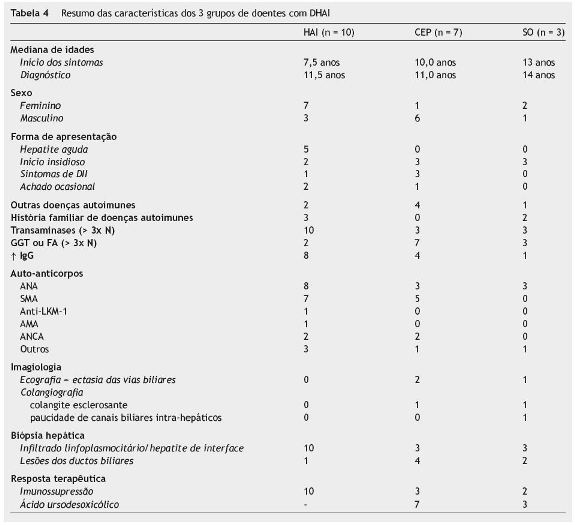
O melhor exame para identificação de CEP é a colangiografia3,14,34,35. A colangioRM é a técnica que deve ser utilizada, por se tratar de um método não invasivo que permite a visualização e caracterização dos ductos biliares intrahepáticos de 3.a e 4.a ordem. A CPRE está atualmente em desuso pelo risco de complicações como pancreatite aguda e colangite4,35, observadas em 2 doentes. A imagem típica da CEP inclui irregularidade dos ductos intra e/ou extra-hepáticos, dilatações saculares focais, aumento do diâmetro do canal biliar comum4,35. Na amostra estudada, foi efetuada colangiografia em 6 doentes (4 com CEP e em 2 com SO). Foram efetuadas 4 CPRE e 2 ColangioRM (exames mais recentes), tendo-se detetado sinais sugestivos de colangite esclerosante em apenas 3 casos. Dos 3 doentes com colangiografia normal, apenas um apresentava lesão ductular no exame histológico. Estes 3 casos correspondem provavelmente a CEP de pequenos ductos.
Histologia hepática
O diagnóstico de HAI implica sempre realização de biópsia hepática4,6. Apesar de haver características histológicas típicas desta patologia (como hepatite de interface e inflamação portal - figura 4), estas não são específicas da doença, podendo ocorrer noutro tipo de hepatopatia6. Outros aspectos histológicos, como o envolvimento dos ductos biliares (ductopenia, colangite), podem ocorrer em 24-31% das crianças com HAI6,30, como observado em um doente.
Embora o exame histológico seja dispensável para o diagnóstico de CEP de grandes ductos quando há evidência de alterações colangiográficas, nos casos de CEP de pequenos ductos é necessária realização de biópsia6,34,35. Os aspetos histológicos mais típicos de CEP incluem inflamação dos espaços porta com infiltrado de linfócitos nos ductos biliares, proliferação e/ou fibrose ductular5,6,8,34,35 - figuras 5 e 6. Em 2 doentes (29%) com CEP e em 1 com SO não se detetaram alterações ductulares no exame histológico, o que pode acontecer sobretudo numa fase inicial da doença4-6,30. Além disso, nos doentes com CEP podem ser observadas caraterísticas histológicas mais sugestivas de HAI, como hepatite de interface6,34,35 - figura 6.
Resposta terapêutica
A evidência de melhoria clínica e laboratorial, após tratamento com corticóides, é um dos critérios de diagnóstico de HAI10,39 e permitiu afirmar o diagnóstico definitivo de HAI nos 3 doentes cujo diagnóstico pré-tratamento era apenas provável.
Nos casos de CEP, verifica-se uma melhor resposta ao tratamento com AUDC8, embora em alguns casos também ocorra melhoria com tratamento imunossupressor5-7. Dos 7 casos de CEP, foi efetuado tratamento imunossupressor em 4, não tendo havido resposta em um, e todos efetuaram depois tratamento com AUDC, com boa resposta.
Nos casos de SO o tipo de resposta à terapêutica (imunossupressão ou AUDC), ou uma alteração dessa resposta ao longo da evolução da doença (caso 19) contribuiu para a suspeita de overlap.
Diagnóstico diferencial com outras patologias
É necessária a exclusão de outras causas de hepatopatia crónica, tais como esteatohepatite não-alcoólica, défice de _-1-antitripsina, doença de Wilson, infeções víricas, álcool e outros tóxicos10. Em todos os doentes a exclusão destas patologias foi possível e de fácil execução.
No caso 5, a presença de ANA, anti-dsDNA e SSA positivos obrigou ao diagnóstico diferencial com lúpus eritematoso sistémico (LES), ilustrando as dificuldades por vezes encontradas na diferenciação entre HAI como entidade independente e o LES com envolvimento hepático40-42. Apesar de a disfunção hepática não estar incluída nos critérios de diagnóstico do LES43, ela pode surgir em cerca de 25-50% dos doentes42,44-46. A patogénese é variável, podendo estar envolvidos diversos mecanismos, entre os quais HAI como uma forma de resposta AI sistémica40,41,44-47. Um estudo inglês evidenciou que a prevalência de HAI nos doentes com LES juvenil era de 9,8%48. Tanto a HAI como o LES estão associados a sinais de autoimunidade como poliartralgia, hipergamaglobulinemia e ANA e dsDNA positivos e ambos respondem favoravelmente aos corticóides40,44,45. Contudo, existem algumas diferenças entre as duas entidades. Em termos histológicos, a HAI caracteriza-se por hepatite de interface, com ou sem envolvimento lobular, e infiltrado linfóide, enquanto no LES a inflamação localiza-se predominantemente a nível lobular e ocasionalmente periportal, com paucidade de infiltração linfóide44,45. Os SMA estão presentes em 60-80% dos doentes com HAI, e em apenas 30% dos doentes com LES, para além de ser possível detetar outros Acs específicos de fígado na HAI45-48. Além disso, a ocorrência de CU pode associar-se a HAI, sendo muito rara a associação com LES45. No caso 5, as características histológicas, a evidência de SMA positivos e a ausência de outras manifestações sugestivas de LES foram aspetos a favor do diagnóstico de HAI. De qualquer forma, a HAI pode surgir anos antes do diagnóstico de LES17,45,48, pelo que deverá ser mantida vigilância nesta doente e efetuada investigação complementar à mínima suspeita de LES.
Diagnóstico diferencial entre HAI, CEP ou SO
A partilha de características clínicas e laboratoriais semelhantes tornam a distinção entre HAI e CEP por vezes difícil - tabela 4. Existem, no entanto, alguns aspetos mais sugestivos de CEP que podem facilitar esta diferenciação: sexo masculino, antecedentes de DII, presença de prurido, curso da doença mais indolente, elevação preferencial da GGT e FA, alterações dos ductos biliares na colangioRM e no exame histológico e melhoria clínica e laboratorial após tratamento com AUCD – tabela 4. Cerca de 45% das crianças com CEP têm DII associada, comparativamente com cerca de 20% das que têm HAI clássica4. Na amostra estudada, esta diferença foi ligeiramente maior (CEP - 57%, HAI - 10%). O tipo de auto-Acs detetados nos 2 tipos de DHAI é semelhante. A exceção parece ser feita no que diz respeito aos ANCA que predominam nos casos de CEP (74 para 56%)4,7,30,35. Na amostra estudada, esta diferença foi inferior (29 para 20%). As alterações ductulares no exame histológico são mais características da CEP, mas podem ocorrer também nas formas de HAI e podem estar ausentes em alguns casos de CEP35, como observado na amostra estudada.
A síndrome de overlap HAI/CEP na criança parece ter uma prevalência semelhante à da HAI4,6. Um estudo de 55 crianças com HAI clássica que realizaram colangiografia, na altura do início da sintomatologia, mostrou que 49% tinham alterações dos ductos biliares característicos de colangite esclerosante, tendo assim sido classificados como SO5,6,30. Na série apresentada não foi efetuada colangiografia em todos os doentes, pelo que o diagnóstico de CEP, e consequentemente de SO, pode ter sido subestimado. Da mesma forma, doentes com CEP podem apresentar, simultaneamente ou posteriormente ao longo da evolução da doença, características de HAI5,30. Num estudo prospetivo de crianças com CEP, verificou-se que 35% vieram a cumprir critérios de HAI6. Na série apresentada, o caso n.◦ 19 exemplifica esta situação. O IAIHG sugere que os doentes com DHAI devem ser classificados em HAI ou CEP, de acordo com o tipo de características que predominam, e que a SO não deve ser considerada como uma entidade independente6.
Conclusões
As maiores dificuldades de diagnóstico foram encontradas nos doentes portadores de CEP com doença em estadio inicial, pela ausência de alterações imagiológicas no colangiograma e pela evidência de anomalias histológicas inespecíficas. Destacam-se as dificuldades colocadas pelos casos de SO, sobretudo aqueles com apresentação sequencial. Salientam-se ainda as dificuldades em efetuar o diagnóstico diferencial entre HAI como entidade independente e outras doenças AI multissistémicas com atingimento hepático, como o LES.
Bibliografia
1. Squires R. Autoimmune hepatitis in children. Ped Gastroenterol. 2004;6:225-30. [ Links ]
2. Gregorio GV, Portmann B, Reid F, Donaldson PT, Doherty DG, McCartney M, et al. Autoimmune hepatitis in childhood: a 20- year experience. Hepatology. 1997;25:541-7. [ Links ]
3. Shneider BL, Suchy FJ. Autoimmune and chronic hepatitis. In: Kliegman RM, Behrman RE, Jenson HB, Stanton BF, editores Nelson Textbook of Pediatrics. 18th ed. Philadelphia: Saunders Elsevier; 2007. p. 1698-701. [ Links ]
4. Mieli-Vergani G, Vergani D. Autoimmune liver disease. In: Deirdre K, editor. Diseases of the liver and biliary system in children. 3rd ed. Oxford: Blackwell publishing;; 2008. p. 191-203. [ Links ]
5. Schramm C, Lohse A. Overlap syndromes of cholestatic liver disease and auto-immune hepatitis. Clin Rev Allergy Immunol. 2005;28:105-14. [ Links ]
6. Boberg KM, Chapman RW, Hirschfield GM, Lohse AW, Manns MP, Schrumpf E, International Autoimmune Hepatitis Group. Overlap syndromes: the international autoimmune hepatitis group (IAIHG) position statement on a controversial issue. J Hepatol. 2011;54:374-85. [ Links ]
7. Invernizzi P, Mackay I. Overlap syndromes among autoimmune liver disease. World J Gastroenterol. 2008;14:3368-73. [ Links ]
8. Maggiore G, Riva S, Sciveres M. Autoimmune disease of the liver and biliary tract and overlap syndromes in childhood. Minerva Gastroenterol Dietol. 2009;55:53-70. [ Links ]
9. Johnson PJ, McFarlane IG. Meeting report: International autoimmune hepatitis group. Hepatology. 1993;18:998-1005. [ Links ]
10. Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, et al. International autoimmune hepatitis group report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929-38. [ Links ]
11. Hennes EM, Zeniya M, Czaja AJ, Parés A, Dalekos GN, Krawitt EL, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology. 2008;48:169-76. [ Links ]
12. Mieli-Vergani G, Vergani D. Autoimmune hepatitis in children: what is different from adult AIH? Semin Liver Dis. 2009;29:297-306. [ Links ]
13. Ramalho H, Santos Silva E. Hepatite autoimune na criança --- revisão de cinco casos. Nascer e Crescer. 2001;10:27-31. [ Links ]
14. Saadah Omar, Smith Arnold, Hardikar Winita. Outcome of autoimmune hepatitis in children --- long-term outcome of autoimmune hepatitis in children. J Gastroenterol Hepatol. 2001;16:1297-302. [ Links ]
15. Ferreira A, Roquete M, Penna F, Toppa N. Hepatite auto-imune em crianças e adolescentes: estudo clínico, diagnóstico e resposta terapêutica. Jornal de Pediatria. 2002;78:309-14. [ Links ]
16. Rafeey M, Kianrad M, Hasani A. Autoimmune hepatitis in iranian children. Indian J Gastroenterol. 2007;26:11-3. [ Links ]
17. Yeh SH, Ni YH, Jeng YM, Chen HL, Wu JF, Chang MH. Pediatr Neonatol. 2009;50:65-9. [ Links ]
18. Ebbeson RL, Schreiber RA. Diagnosing autoimmune hepatitis in children: is the International Autoimmune Hepatitis Group scoring system useful? Clin Gastroenterol Hepatol. 2004;2:935-40. [ Links ]
19. Mieli-Vergani G, Vergani D. Autoimmune hepatitis in children. Clin Liver Dis. 2001;6:335-46. [ Links ]
20. Baranov AA, Kaganov BS, Gundobina OS, Zainudinov ZM. Autoimmune hepatitis in children. Intern Pediatr. 2003;18:23-9. [ Links ]
21. Debray D, Maggiore G, Bernard O. Cyclosporin treatment of autoimmune hepatitis (AIH) in children. J Pediatr Gastroenterol Nutr. 1995;20:470. [ Links ]
22. Mieli-Vergani G, Vergani D. Progress in pediatric autoimmune hepatitis. Semin Liver Dis. 1994;14:282-8. [ Links ]
23. Fainboim L, Marcos Y, Pando M, Capucchio M, Reyes GB, Galoppo C, et al. Chronic active immune hepatitis in children. Strong association with a particular HLA-DR6 (DRB1*1301) haplotype. Hum Immunol. 1994;41:146-50. [ Links ]
24. Maggiore G, Veber F, Bernard O, Hadchouel M, Homberg JC, Alvarez F, et al. Autoimmune hepatitis associated with anti-actin antibodies in children and adolescents. J Pediatr Gastroenterol Nutr. 1993;17:376-81. [ Links ]
25. Codoner-Franch P, Bernard O, Maggiore G, Alagille D, Alvarez F. Clinical and imunnological heterogeneity of anti-liver-kidney microssome antibody-positive autoimmune hepatitis in children. J Pediatr Gastroenterol Nutr. 1989;9:436-40. [ Links ]
26. Maggiore G, Bernard O, Hadchouel M, Hadchouel P, Odievre M, Alagille D. Treatment of autoimmune chronic active hepatitis in childhood. J Pediatr. 1984;104:839-44. [ Links ]
27. Yachha SK, Srivstava A, Chetri K, Saraswat VA, Krishnani N. Autoimmune liver disease in children. J Gastroenterol Hepatol. 2001;16:674-7. [ Links ]
28. Mieli-Vergani G, Vergani D. Autoimmune liver disease in children. Ann Acad Med Singapore. 2003;32:239-43. [ Links ]
29. Maggiore G, Bernard O, Homberg JC, Hadchouel M, Alvarez F, Hadchouel P, et al. Liver disease associated with antiliver-kidney microsome antibody in children. J Pediatr. 1985;108:399-404. [ Links ]
30. Gregorio GV, Portmann B, Karani J, Harrison P, Donaldson PT, Vergani D, et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: a 16-year prospective study. Hepatology. 2001;33:544-53. [ Links ]
31. Oliveira D, Teixeira P, Silva ES, Silvestre F, Medina M. Um caso de síndrome variante de doença hepática autoimune. Acta Pediatr Port. 2000;31:167-71. [ Links ]
32. Gopal S, Nagral A, Mehta S. Autoimmune sclerosing cholangitis: an overlap syndrome in a child. Indian J Gastroenterol. 1999;18:31-2. [ Links ]
33. El-Shabrawi M, Wilkinson ML, Portmann B, Mieli-Vergani G, Chong SK, Williams R, et al. Primary sclerosing cholangitis in childhood. Gastroenterology. 1987;92:1226-35. [ Links ]
34. Superina R. Biliary disease in infants and children. In: Clavien PA, Baillie J, editores Diseases of the gallbladder and bile ducts - diagnosis and treatment. 2nd ed. Oxford: Blackwell publishing; 2006. p. 377-410. [ Links ]
35. EASL, Clinical Practice Guidelines.Management of cholestatic liver diseases. J Hepatology. 2009;51:237-67. [ Links ]
36. Czaja AJ. Behavior and significance of autoantibodies in type 1 autoimmune hepatitis. J Hepatol. 1999;30:394-401. [ Links ]
37. Clifford BD, Donahue D, Smith L, Cable E, Luttig B, Manns M, et al. High prevalence of serological markers of autoimmunity in patients with chronic hepatitis C. Hepatology. 1995;21: 613-9. [ Links ]
38. Muratori L, Parola M, Ripalti A, Robino G, Muratori P, Bellomo G, et al. Liver/kidney microsomal antibody type 1 and liver cytosol antibody type 1 concentrations in type 2 autoimmune hepatitis. Gut. 1998;42:721-6. [ Links ]
39. Czaja AJ, Freese DK. Diagnosis and treatment of autoimmune hepatitis. Hepatology. 2002;36:479-97. [ Links ]
40. Youssef WT, Tavill AS. Connective tissue disease and the liver. J Clin Gastroenterol. 2002;35:345-9. [ Links ]
41. Fonseca JE, Reis P, Saraiva F, Crujo C, Baptista A, da Silva JA, et al. A complex case of hepatitis in a patient with systemic lupus erythematosus. Clin Rheumatol. 1999;18:414-6. [ Links ]
42. Islam S, Riordan Jw, Mcdonald JA. A rare association of primary biliary cirrhosis and systemic lupus erythematosus and review of the literature. J Gastroenterol Hepatol. 1999;14:431-5. [ Links ]
43. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725. [ Links ]
44. Schiavon LL, Carvalho-Filho RJ, Narciso-Schiavon JL, Lanzoni VP, Ferraz ML, Silva AE. Late-onset systemic lupus erythematosusassociated liver disease. Rheumatol Int. 2010;30:1515-8. [ Links ]
45. Yamasaki S, Origuchi T, Nakata K, Toriyama K, Taguchi T, Ida H, et al. Autoimmune hepatitis in a patient with systemic lupus erythematosus: a case report. Mod Rheumatol. 2004;14:169-73. [ Links ]
46. Van Hoek B. The spectrum of liver disease in systemic lupus erythematosus. Neth J Med. 1996;48:244-53. [ Links ]
47. Chowdhary VR, Crowson CS, Poterucha JJ, Moder KG. Liver involvement in systemic lupus erythematosus: a case review of 40 patients. J Rheumatol. 2008;35:2159-64. [ Links ]
48. Irving KS, Sen D, Tahir H, Pilkington C, Isenberg DA. A comparison of autoimmune liver disease in juvenile and adult populations with systemic lupus erythematosus - a retrospective review of cases. Rheumatol. 2007;46:1171-3. [ Links ]
Conflito de interesses
Os autores declaram não haver conflito de interesses.
*Autor para correspondência
Correio eletrónico: martariospinho@gmail.com (M. Rios).
Recebido a 16 de setembro de 2011; aceite a 9 de janeiro de 2012
Abreviaturas:
Acs, anticorpos; AI, autoimune; AMA, anticorpos antimitocondriais; ANA, anticorpos antinucleares; ALT, alanina aminotransferase; anti-LC1, anti-citosol hepático tipo 1; AST, aspartato aminotransferase; AUDC, ácido ursodesoxicólico; ANCAs, anticorpos anticitoplasma dos neutrófilos; BT, bilirrubina total; BD, bilirrubina direta; CBP, cirrose biliar primária; CEP, colangite esclerosante primária; CMV, citomegalovírus; CPRE, colangiopancreatografia retrógrada endoscópica; CU, colite ulcerosa; DII, doença inflamatória intestinal; DHAI, doença hepática autoimune; EBV, epstein-barr vírus; FA, fosfatase alcalina; GGT, gama-glutamil-transferase; HAI, hepatite autoimune; LES, lúpus eritematoso sistémico; PR3, anti-proteinase 3; SO, síndrome de overlap; SMA, anticorpos anti-músculo liso; TC, tomografia computorizada.
