










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkJornal Português de Gastrenterologia
versão impressa ISSN 0872-8178
J Port Gastrenterol. vol.18 no.6 Lisboa nov. 2011
Diarreia Intratável: Doença de Inclusão das Microvilosidades num Recém-Nascido
Isabel Valente1, Rosa Lima1, Carmen Carvalho1, Helena Ferreira1, Fernanda Marcelino1, Céu Mota1, Ana Ramos1, Carlos Duarte1, Fátima Carneiro2, Fernando Pereira1, Herculano Rocha1
1Departamento de Pediatria. Centro Hospitalar do Porto – Unidade Hospital de Crianças Maria Pia, Porto - Portugal; 2Serviço de Anatomia Patológica, Hospital de S. João, Faculdade de Medicina da Universidade do Porto e IPATIMUP – Portugal
RESUMO
Os defeitos congénitos da mucosa intestinal podem provocar diarreia severa no período neonatal com compromisso nutricional importante e consequentemente dependência da nutrição parentérica. Neste trabalho apresentamos o caso de um recém-nascido do sexo feminino, com diarreia líquida intratável, desidratação grave e acidose metabólica persistente, apesar de pausa alimentar, requerendo nutrição parentérica. Após uma investigação exaustiva incluindo estudo microbiológico, imunológico e metabólico, o diagnóstico de doença de inclusão das microvilosidades foi feito, com base nas características histológicas e imunocíticas observadas na biopsia duodenal, com a presença de inclusões citoplasmáticas localizadas no pólo apical dos enterócitos e apagamento da bordadura em escova. O transplante intestinal pode ser o único tratamento para este raro distúrbio intestinal congénito.
PALAVRAS-CHAVE: Diarreia intratável; Recém-nascido; Doença de inclusão das microvilosidades.
Intractable Diarrhea: Microvillous Inclusion Desease in a Newborn Infant
ABSTRACT
Congenital defects of the intestinal mucosa cause severe diarrhoea in the neonatal period with important nutritional impact and subsequent dependency on parenteral nutrition. We present a case of a newborn female with protracted watery diarrhoea, severe dehydratation and persistent metabolic acidosis despite bowel rest, requiring parenteral nutrition. After extensive investigation including microbiologic, immunologic and metabolic studies, the diagnosis of microvillous inclusion disease was done, on the basis of histological and immunohistochemical features observed in a duodenal biopsy, showing intracytoplasmatic inclusions in the apical pole of the cytoplasm of enterocytes and effacing of the brush border. Intestinal transplantation may be the only treatment for this rare intestinal disorder.
KEYWORDS: Protracted diarrhoea; Newborn; Microvillous inclusion disease.
INTRODUÇÃO
A diarreia intratável é actualmente definida como uma perda persistente e frequente de grande volume de fezes, cuja gravidade requer um suporte nutricional, frequentemente na forma de nutrição parentérica1.
As causas incluem enteropatias infecciosas e pós-infecciosas, síndrome do intestino curto, distúrbios graves da motilidade (aganglionismo total ou subtotal, síndrome de pseudo-obstrução intestinal crónico) e doenças congénitas do desenvolvimento do enterócito2.
A doença de inclusão das microvilosidades (DIM) é um distúrbio congénito raro das células epiteliais intestinais que, na sua forma típica, se apresenta inicialmente como uma diarreia líquida intratável no período neonatal que pode ser ameaçadora da vida e que é caracterizado por anomalias morfológicas típicas dos enterócitos2, reflectindo uma incapacidade para transportar nutrientes simples associado a uma secreção aumentada para o lúmen intestinal3.
A DIM manifesta-se nos primeiros dias de vida (forma de aparecimento precoce) ou nos primeiros dois meses de vida (forma de aparecimento tardio). É uma doença rara, de transmissão hereditária autossómica recessiva4.
O diagnóstico definitivo é feito com base nas alterações morfológicas, identificáveis por estudo histológico, imunocitoquímico e ultrastrutural, sendo característica a presença de inclusões intracitoplasmáticas revestidas por microvilosidades, localizadas no polo apical dos enterócitos5.
CASO CLÍNICO
Recém-nascido do sexo feminino, primeiro filho de pais jovens, saudáveis, não consanguíneos. Gestação vigiada, de 35 semanas. Serologias maternas à data do parto (VDRL, AgHbs, Ac anti – HIV - 1 e 2) negativas. Parto por cesariana, com Índice de Apgar de 9/10 ao primeiro e quinto minuto respectivamente. Somatometria adequada à idade gestacional (peso 2545 gr, comprimento 46 cm, perímetro cefálico 33 cm) e exame objectivo ao nascimento sem particularidades. A primeira eliminação de mecónio decorreu dentro das primeiras 48 horas. Iniciou alimentação com leite hipoalergénico, por hipogalactea materna, tendo alta hospitalar às 72 horas de vida. Esteve clinicamente bem até ao 5º dia de vida altura em que iniciou quadro de recusa alimentar, irritabilidade e diarreia líquida. No dia seguinte foi admitido no Hospital da área por desidratação grave (perda ponderal de 25%), acidose metabólica (pH: 6,9; bicarbonatos: 5 mmol/L; base excesso: - 28,7 mmol/L) e insuficiência renal (creatinina: 2,14 mg/dl; ureia: 93 mg/dl). Foi efectuado rastreio infeccioso e apesar de negativo foram instituídas antibioterapia endovenosa e fluidoterapia com suplemento de bicarbonato de sódio endovenoso.
Foi transferido ao 7º dia de vida para o Hospital Maria Pia, apresentando-se na admissão irritável, sem dismorfias aparentes, com peso de 2000 gr, hemodinamicamente estável, polipneico, fontanela anterior deprimida, pele ictérica e seca, mucosas secas com tempo de preenchimento capilar inferior a 2 segundos, com o restante exame objectivo sem alterações. Repetiu rastreio analítico que evidenciou: hemoglobina - 17,2 g/dl; hematócrito - 53,2%; leucócitos - 34250/mm3, com 64,5% de neutrófilos, 21,8% de linfócitos e 1,3% de eosinófilos; plaquetas - 71 3000/mm3; proteína C reactiva (PCR) - 0,02 mg/dl. A bioquímica sérica evidenciou: creatinina -134 mmol/L; ureia - 18 mmol/L; bilirrubina total - 157 μmol/L; TGP - 37 U/L; sódio - 153 mmol/L; potássio - 4,9 mmol/L; cloro - 114 mmol/L; cálcio - 2,28 mmol/L; fósforo - 1,63 mmol/L; magnésio - 0,97 mmol/L; pH - 7,25; bicarbonatos - 7,5 mmol/L; base excesso - 19,6 mmol/L. A telerradiografia póstero-anterior do tórax e a radiografia abdominal não mostraram alterações.
Após correcção do desequilíbrio hidroelectrolítico e metabólico, mantendo-se em pausa alimentar, a diarreia persistia com débitos fecais de 60 – 100 ml/Kg/dia, tendo iniciado nutrição parentérica total por perda ponderal significativa.
Ao 10º dia de vida reiniciou alimentação entérica com dieta hipoproteica, com proteínas inteiras e lactose, por suspeita de doença do metabolismo dos aminoácidos, tendo ocorrido um agravamento da diarreia, perda ponderal e acidose metabólica. A pesquisada de substâncias redutoras nas fezes foi positiva.
Foram colocadas as seguintes hipóteses de diagnóstico: enteropatia alérgica, defeitos congénitos de absorção de carbo-hidratos, defeitos congénitos de transporte de iões, doença metabólica, enteropatia infecciosa e fibrose quística. Realizaram-se novos exames complementares de diagnóstico incluindo hemocultura, coprocultura, exame virulógico de fezes, urocultura, pesquisa de leucócitos fecais e o RAST para as proteínas do leite de vaca, tendo sido todos negativos. O ionograma fecal revelou os seguintes resultados: sódio - 83 mEq/L, potássio - 18,5 mEq/L, cloro - 78 mEq/L; osmolaridade fecal - 253 mEq/L (normal; 280 - 320 mEq/L), com gap osmótico de 43,5 mOsm/Kg, sugerindo diarreia secretora. O estudo metabólico, ecografia transfontanelar, abdominal, renal, pélvica e cardíaca foram normais e a tripsina imunorreactiva negativa. O doseamento de imunoglobulinas e níveis de complemento foram normais. As serologias (sífilis, hepatite, CMV, HIV 1 e 2) foram negativas.
Ao 28º dia de vida foi realizada endoscopia digestiva alta com biopsia duodenal que evidenciou duas úlceras gástricas na grande curvatura, com características de úlceras de stress. O resultado do exame histológico da biopsia duodenal não permitiu avaliar adequadamente a morfologia vilositária, por deficiente inclusão do fragmento, tendo-se observado infiltrado linfo-plasmocitário no córion da mucosa.
Após introdução gradual da alimentação (incluindo dieta semi-elementar, elementar, isenta de glicose e galactose) verificou-se agravamento clínico com reinício de diarreia, acidose metabólica, diminuição do pH fecal e presença de açúcares redutores nas fezes, compatível com quadro de diarreia do tipo osmótico. Nos períodos de pausa alimentar o ionograma fecal evidenciava um aumento do sódio, sem gap osmótico, a favor de uma diarreia do tipo secretora (Fig.1).
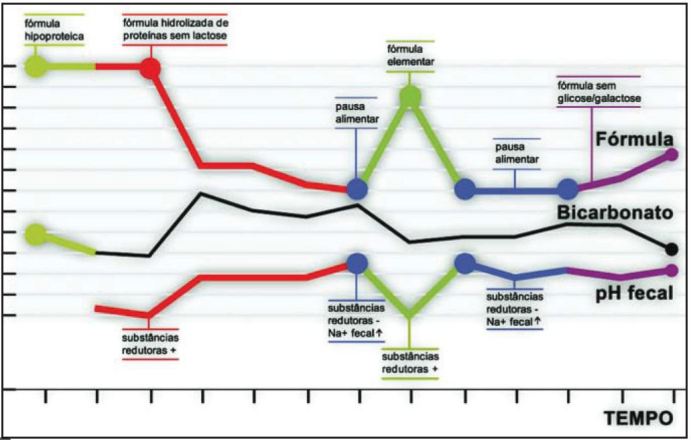
Fig. 1. Gráfico da variação do pH fecal e bicarbonato sérico em função do tipo de fórmula alimentar utilizada.
Foram então colocadas novas hipóteses de diagnóstico: enteropatia autoimune, displasia intestinal epitelial (tufting enteropathy) e doença de inclusão das microvilosidades (DIM).
A biopsia duodenal e rectal foi repetida ao 78º dia. Na primeira, observaram-se vilosidades intestinais alargadas e encurtadas com infiltrado inflamatório discreto no córion e epitélio de revestimento colunar com microvacuolização do citoplasma apical (Fig. 2A); a técnica histoquímica de PAS (Periodic Acid-Schiff) após diastase evidenciou bordadura em escova mal definida e presença de pequenos vacúolos no citoplasma apical dos enterócitos (Fig. 2B); no estudo imunocitoquímico observou-se imunoexpressão de CD10 nos vacúolos citoplasmáticos apicais (Fig. 2C). Estes aspectos, no contexto clínico anteriormente descrito, permitiram o diagnóstico de Doença de Inclusão das Microvilosidades.
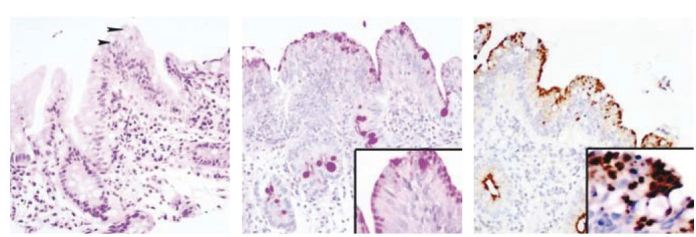
Fig. 2. Aspecto histológico, histoquímico e imunocitoquímico da biopsia duodenal realizada ao 78º dia: A) Aspecto histológico, com encurtamento e alargamento das vilosidades (sem hiperplasia das criptas) e microvacuolização do citoplasma apical (pontas de setas). B) Presença de inclusões no citoplasma apical dos enterócitos realçadas pela técnica histoquímica de PAS após diastase; Inset: maior ampliação de uma das vilosidades intestinais, evidenciando as inclusões PAS-positivas. C) Imunoexpressão de CD10 nas inclusões citoplasmáticas apicais; Inset: maior ampliação do revestimento de uma das vilosidades intestinais, realçando as inclusões no citoplasma apical dos enterócitos e ausência de bordadura em escova.
O exame histológico da biopsia rectal não apresentava alterações.
A criança manteve-se em nutrição parentérica total com suplementos de bicarbonato de sódio, havendo melhoria clínica, aumento ponderal, diminuição dos episódios de diarreia e normalização do equilíbrio ácido-base. Os análogos da somatostatina (octreotido) foram experimentados mas sem eficácia clínica.
A doente foi transferida para o Hospital Necker-Enfants Malades – Paris para programação de Transplante Intestinal.
DISCUSSÃO
As causas de diarreia neonatal podem ser divididas em adquiridas ou congénitas e, de acordo com a arquitectura vilositária, em entidades com arquitectura vilositária normal (incluindo a diarreia secretora com perda de cloro ou de sódio; defeitos congénitos de absorção de carbo-hidratos; má absorção de ácidos biliares; intestino curto congénito) ou com atrofia vilositária (DIM, enteropatia com tufos (tufting enteropathy); enteropatia autoimune; enteropatia alérgica, enteropatia infecciosa e pós-infecciosa)1.
Embora rara, a DIM deve ser considerada no diagnóstico diferencial da diarreia crónica intratável do lactente.
No nosso caso clínico a diarreia apresentou-se clínicamente como uma diarreia do tipo secretor, uma vez que nos períodos de pausa alimentar o ionograma fecal evidenciava um aumento do sódio, sem gap osmótico (Fig.1). Com a introdução da alimentação verificava-se agravamento clínico com reinício de diarreia, diminuição do pH fecal e presença de açúcares redutores nas fezes, aspectos compativeis com quadro de diarreia do tipo osmótico. Portanto, a existência de diarreia, de início no período neonatal, com um componente misto é a favor de uma enteropatia com atingimento do eixo vilositário como a enteropatia autoimune, a designada tufting enteropathy ou a doença de inclusão das microvilosidades (DIM). O exame histológico permite o diagnóstico diferencial entre estas entidades1,2.
A DIM na sua forma típica apresenta-se como uma diarreia líquida grave manifestando-se nas primeiras horas ou dias de vida; a diarreia é do tipo secretor, com volumes fecais compreendidos entre 50 – 300 ml/Kg/dia, apesar de pausa alimentar, com concentrações dos electrólitos fecais aumentadas, sem gap osmótico. O reinício da alimentação induz uma diarreia osmótica com aumento do volume fecal (100-500 ml/Kg/dia) devido à alteração da estrutura das microvilosidades, com inclusão das mesmas em cistos localizados no citoplasma apical, assim como atrofia de grau variável das vilosidades intestinais6.
É uma doença de transmissão autossómica recessiva, com um predomínio no sexo feminino; contrariamente a outras diarreias congénitas secretoras, a DIM não se acompanha de poli-hidrâmnios6. A patogénese ainda não está totalmente esclarecida, Phillips et al sugeriram que a DIM se associa a um defeito na exocitose do glicocálice7. Recentemente, mutações em genes (MYO5B e RAB8) que codificam para proteínas conhecidas por estar envolvidas no transporte apical/basolateral, em células epiteliais polarizadas, foram encontradas em famílias de doentes com DIM, sugerindo nalguns casos uma transmissão digénica. No entanto o espectro de mutações possíveis ainda não está claramente definido8-10.
O diagnóstico pode ser estabelecido por biopsia duodenal ou jejunal com identificação das características histológicas e imunocitoquímicas anteriormente descritas. A microscopia electrónica pode também dar um contributo importante para o diagnóstico, através da identificação de cistos citoplasmáticos apicais revestidos por microvilosidades5,6,11.
O prognóstico é reservado. Um estudo multicêntrico de 23 crianças com DIM revelou uma expectativa de vida reduzida, com uma taxa de sobrevivência ao primeiro ano inferior a 25%12,13; no caso de DIM de aparecimento tardio o prognóstico é mais favorável estando descrita sobrevivência mais longa com nutrição parentérica parcial2,14.
A nutrição parentérica total (NPT) e o transplante intestinal ou combinado (intestinal-hepático, quando se desenvolve insuficiência hepática secundária) são o único tratamento para a DIM.
A NPT de longo termo na maioria dos lactentes pode aumentar o risco de complicações relacionadas com o catéter (sepsis, trombose) e doença hepática, pelo que o transplante isolado de intestino delgado ou intestinal e hepático constitui uma alternativa terapêutica, com uma taxa de sobrevivência aos 5 anos de aproximadamente 50%13,15.
Este relato clínico salienta a importância da realização precoce de uma biopsia intestinal (duodenal ou jejunal) como um método relativamente simples que permite o diagnóstico precoce num recém-nascido com diarreia persistente, após exclusão das causas mais frequentes através de métodos de investigação não invasivos. O diagnóstico é muitas vezes atrasado devido à dificuldade em obter-se uma biopsia de intestino delgado no período neonatal. Dado que a evidência científica aponta para uma base genética da DIM, o aconselhamento genético nas famílias afectadas é essencial.
REFERÊNCIAS
1. Sherman PM, Mitchell DJ, Cutz E. Neonatal enteropathies: defining the causes of protracted diarrhea of infancy. J Pediatr Gastroenterol Nutr 2004;38:16-26. [ Links ]
2. Goulet O. Congenital Enteropathy and Intestinal Transplantation. Ann Nestlé 2006;64:25-37. [ Links ]
3. Davidson GP, Cutz E, Hamilton JR. Familial enteropathy: A syndrome of protracted diarrhea from birth, failure thrive and, hypoplastic villus atrophy. Gastroenterology 1978;75:783-90. [ Links ]
4. Ruemmele FM, Schmitz J, Goulet O. Microvillous inclusion disease (microvillous atrophy). Orphanet J Rare Dis 2006;1:22. [ Links ]
5. Bell SW, Kerner JA Jr, Sibley RK. Microvillous inclusion disease. The importance of electron microscopy for diagnosis. Am J Surg Pathol 1991;15:1157-1164. [ Links ]
6. Guandalini S, Nocerino A. Congenital microvillus atrophy. Available at: URL: http://www.emedicine.com/ped/topic461.htm. (Acesso em Setembro 2011). [ Links ]
7. Phillips AD, Brown A, Hicks S, et al. Acetylated sialic acid residues and blood group antigens localise within the epithelium in microvillous atrphy indicating internal accumulation of the glycocalyx. Gut 2004;53:1764-1771. [ Links ]
8. Sato T, Mushiake S, Kato Y, et al. The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature 2007;448:366-369. [ Links ]
9. Müller T, Hess M, Schiefermeier N, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008;40:1163-1165. [ Links ]
10. Erickson R, Larson-Thomé K, Valenzuela R K, et al. Navajo microvillous inclusion disease is due to a mutation in MYO5B. Am J Med Genet A 2008;24:3117-3119. [ Links ]
11. Beck NS, Chang YS, Kang IS, et al. Microvillus inclusion disease in two Korean infants. J Korean Med Sci 1997;12:452-456. [ Links ]
12. Phillips AD, Schmitz J. Familial microvillous atrophy: a clinicopathological survey of 23 cases. J Pediatr Gastroenterol Nutr 1992;14:380-396. [ Links ]
13. Goulet O, Ruemmele F, Lacaille F, et al. Irreversible intestinal failure. J Pediatr Gastroenterol Nutr 2004;38:250-269. [ Links ]
14. Kučinskienė R, Jančiauskas D, Puas A, et al. Microvillous inclusion disease. Medicina (Kaunas) 2004;40:864-867. [ Links ]
15. Pecache N, Patole S, Hagan R, et al. Neonatal congenital microvillus atrophy. Postgraduate Medical Journal 2004;80:80-83. [ Links ]
16. Cutz E, Rhoads JM, Drumm B et al. Microvillus inclusion disease: an inherited defect of brush-border assembly and differentiation. N Engl J Med 1989;320:646-651. [ Links ]
17. Goulet O, Kedinger M, Brousse N, et al. Intractable diarrhea of infancy with epithelial and basement membrane abnormalities. J Pediatr 1995;127:212-219. [ Links ]
18. Weeks DA, Zuppan CW, Malott RL, et al. Microvillous inclusion disease with abundant vermiform, electron-lucent vesicles. Ultrastruct Pathol 2003;27:337-340. [ Links ]
19. Wilson W, Scott RB, Pinto A, et al. Intractable diarrhea in a newborn infant: microvillous inclusion disease. Can J Gastroenterol 2001;15:61-64. [ Links ]
20. Catassi C, Fabiani E, Spagnuolo MI, et al. Severe and protracted diarrhea: results of the 3-year SIGEP multicenter survey. Working Group of the Italian Society of Pediatric Gastroenterology and Hepatology (SIGEP). J Pediatr Gastroenterol Nutr 1999;29:63-68. [ Links ]
21. Nathavitharana KA, Green NJ, Raafat F, et al. Siblings with microvillous inclusion disease. Arch Dis Child 1994;71:71-73. [ Links ]
22. Ruemmele FM, Jan D, Lacaille F, et al. New perspectives for children with microvillous inclusion disease: early small bowel transplantation. Transplantation 2004;77:1024-1028. [ Links ]
Correspondência: Rosa Maria Lima; Rua da Boavista, 827 - 4050 - 111 Porto - Portugal; Tel: +351 919 531 949; E-mail: rosalima@netcabo.pt;
Recebido para publicação: 07/11/2009 e Aceite para publicação: 19/12/2010
