











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
A osteogénese imperfeita é uma doença do tecido conjuntivo, rara e hereditária com incidência estimada de 1 : 10 000 a 15 0001 nascimentos. É caraterizada principalmente por fragilidade óssea com fraturas recorrentes e manifestações sistémicas com escleras azuis, dentinogénese imperfeita, perda auditiva e laxidão ligamentar. Apresenta grande variedade clínica, desde uma forma letal perinatal até formas atenuadas com menor número de fraturas. A classificação de Sillence et al (1979) descrevia 4 tipos de OI.2 Esta classificação foi posteriormente revista por Warman et al3 (2011 ), que acrescentou outro tipo baseado em critérios clínicos. O tipo I é a forma mais frequente com fraturas usualmente durante a infância e sem grande deformidade óssea; as escleras azuis podem estar presentes, assim como outras manifestações secundárias. O tipo II é a forma letal com fraturas descritas desde as 14-16 semanas. No tipo III observa-se uma alta frequência de fraturas com deformidades acumuladas do esqueleto. No tipo IV a gravidade pode ser variável podendo ser deformante, mas não tanto quanto o tipo III. O tipo V é mais rara com formação de calos hipertróficos nos ossos longos nos locais de fraturas passadas.3•4
Em 90% dos doentes encontram-se mutações dominantes nos genes que codificam as cadeias alfa1 e alfa2 do colagénio tipo 1 (COL1A1 e COL1A2),5 sendo estas as mutações clássicas. As alterações patológicas ocorrem em todos os tecidos em que o colagénio tipo 1 seja um constituinte importante (sobretudo osso, ligamentos, dentina e escleras).
Formas mais ligeiras de doença são observadas com defeitos quantitativos, enquanto que mutações qualitativas levam a doença mais grave, com alterações da matriz extracelular, que é mais suscetível a processos de reparação.6•7Têm sido descritos casos com mutações recessivas e variantes ligadas ao X em doentes com fenótipo característico de OI. 4•8Com o aumento de novas mutações foram sendo revistos os tipos de OI, estando atualmente descrito até ao tipo XIV, sendo que os novos tipos podem corresponder a fenótipos sobreponíveis aos tipos I-V mas com diferentes mutações.4
Não há cura para esta doença e o tratamento de suporte passa por tratamento ortopédico, estomatológico e fisioterapia. Do ponto de vista farmacológico o tratamento consiste em suplementação de cálcio e vitamina D9 e uso de bifosfonatos cujo objetivo é inibir o turnover ósseo aumentando massa óssea, promovendo maior estabilidade e força do osso. No entanto esta terapêutica não atua no defeito primário da 01 e como tal a sua ação pode ficar aquém do desejado. Uma revisão sistemática10 avaliou 14 ensaios clínicos e concluiu que os bifosfonatos aumentam a densidade mineral óssea (DMO), mas não comprovam consistentemente a redução no número de fraturas, a melhoria na redução da dor ou a melhoria da mobilidade.
Material e Métodos
Estudo observacional e retrospetivo com base em dados obtidos através da consulta de processos clínicos de 20 doentes com idade superior a 18 anos e com diagnóstico clínico de OI, seguidos na consulta externa de Doenças Hereditárias do Metabolismo do Centro Hospitalar. Foram registados dados da história médica reportada pelos doentes, do exame físico realizado pelo médico assistente, dos resultados laboratoriais, radiográficos e de densitometrias ósseas, bem como as terapêuticas utilizadas.
A DMO foi medida na coluna lombar e no fémur proximal e expressa em Te Z scores, através da densitometria óssea. Foram excluídos os dados em doentes que apresentam implantes ou material ortopédico na região da medição da DMO. As características clínicas e dados bioquímicos foram apresentados sob a forma de média ± desvio padrão ou sob a forma de proporção em percentagem (%). Os resultados de T-score e Z-score foram apresentados sob a forma de mediana ± amplitude interquartil. Os doentes foram distribuídos por grupos de acordo com a classificação de Sillence et al. Para investigar diferenças entre o género masculino e feminino foram realizados o Teste-T para amostras independentes e o teste U de Mann-Whitney conforme apropriado. O valor de p < 0,05 foi o nível de significância estatístico considerado. Para avaliar a existência de correlação entre o número de fraturas e os valores do T-score ou Z-score para cada tipo de OI foi utilizado o coeficiente de Tau-b de Kendall. Todos os dados foram analisados através do programa informático Statistical Package for the Social Sciences, SPSS, versão 27,0.
Resultados
As caraterísticas clínicas, dados analíticos e de DMO dos doentes estão sumarizadas na Tabela 1.
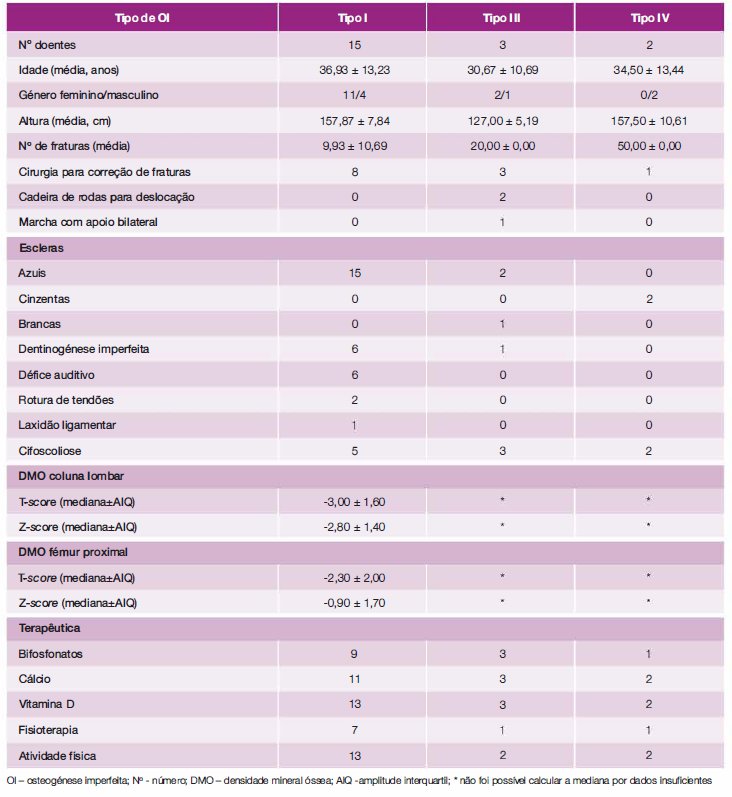
Dos 20 doentes, 65,0% (n = 13) eram do sexo feminino, com idades compreendidas entre os 19 e 61 anos, com apenas três doentes com mais de 50 anos, e média de idade de 35,75 ± 12,44 anos. Relativamente ao tipo de OI, 75,0% dos doentes era do tipo I (n = 15), 15,0% (n = 3) do tipo III e 10,0% (n = 2) do tipo IV. No tipo I e no tipo III a maioria dos doentes era do género feminino, no tipo IV os dois doentes eram do género masculino. A média de alturas no tipo I foi de 157,87 ± 7,84 cm, no tipo III foi de 127,00 ± 5, 19 cm e no tipo IV foi de 157,50 ± 10,61 cm, sendo que a diferença entre a média das alturas entre o tipo I e III e entre o tipo III e o tipo IV foi estatisticamente significativa. O diagnóstico em quatro doentes ocorreu no período neonatal, em dois doentes enquanto lactentes, seis na infância, um na adolescência e sete apenas na idade adulta, apesar de seis destes apresentarem sintomas desde a infância. Em dez doentes (50,0%) foi encontrada mutação no gene COL1A1, num doente no gene COL1A2, em três doentes (15,0%) não foram encontradas mutações e seis (30,0%) ainda não dispunham de teste genético.
Constatou-se um número variável de fraturas por doente que se situou entre 2 e 50. A média de fraturas foi superior nos doentes do tipo IV (50,00 ± 00,00) e do tipo III (20,00 ± 00,00) em relação aos doentes do tipo I (9,93 ± 10,69). Não houve diferença significativa do número médio de fraturas entre género masculino e feminino. Relativamente aos locais mais frequentes de fratura, 55,5% (n = 11) dos doentes apresentou fratura do fémur, 50,0% (n = 10) da coluna (cervical, lombar ou dorsal), 40,0% (n = 8) dos dedos da mão ou pé, 40,0% (n = 8), da tíbia ou perónio e 40,0% (n = 8) do rádio ou cúbito.
odos os doentes do tipo I apresentavam escleras azuis. A dentinogénese imperfeita e o défice auditivo foram descritos em sete e seis doentes, respetivamente. Nenhum doente apresentava envolvimento cardíaco e numa doente foi objetivada síndrome ventilatória restritiva ligeira.
Relativamente à DMO, no tipo Ia mediana do T-score do fémur proximal foi de -2,30 ± 2,00 e da coluna lombar foi de -3,00 ± 1,60, o Z-score do fémur proximal foi de-0,90 ± 1,70 e o da coluna lombar foi de -2,80 ± 1,40 (Tabela 1). Para os restantes tipos de 01 não foi possível o cálculo da mediana dado o número insuficiente de dados de doentes. No caso dos doentes do tipo III encontravam-se disponíveis apenas valores referentes à coluna lombar de dois doentes (T-score de -3,0 e -3,5 e de Z-score de -2,3 e -2,7). No caso dos dois doentes do tipo IV, os valores de T-score do fémur proximal foram de -0,2 e 0,7 e T-score da coluna lombar de -5,2 e -2,7; relativamente aos valores Z-score, estes estavam disponíveis para apenas um doente (Z-score fémur proximal de -0,7 e Z-score da coluna lombar de -2,7). As diferenças das medianas dos T-score e Z-score entre o género masculino e feminino também não foram significativas. Em dois doentes do género feminino não foi realizada densitometria óssea. Não foi encontrada uma correlação estatisticamente significativa entre o número de fraturas e o valor da mediana dos T-score e Z-score nos doentes do tipo I.
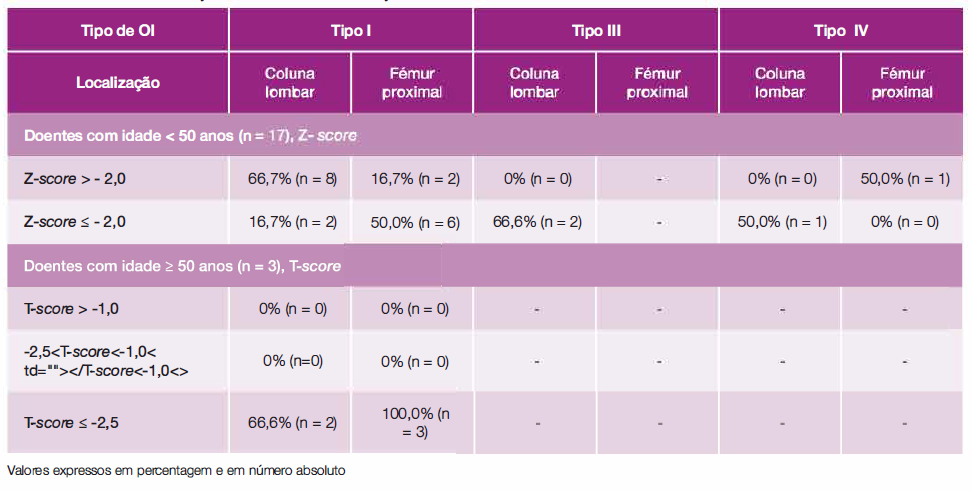
A Tabela 2 apresenta os Z-score para doentes com menos de 50 anos de idade por cada tipo de OI e os T-score para os doentes com mais de 50 anos de acordo com a lnternational Society for Clinical Densitometry. Da análise podemos verificar que 50,0% dos doentes com menos de 50 anos e 100,0% dos doentes com mais de 50 anos do tipo I apresentavam critérios de osteoporose, bem como 66,6% e 50,0% dos doentes do tipo III e IV, respetivamente.
Tabela 2: Z-score para doentes com menos de 50 anos de idade e os T-score para os doentes com mais de 50 anos de acordo com a lnternational Society for Clinical Densitometry
A maioria dos doentes (90,0%) foi medicada com bifosfonatos em algum momento da sua vida. Dos 18 doentes medicados com bifosfonatos, 55,6% (n = 10) foram tratados na vida adulta, 27,8% (n = 5) na infância/adolescência e vida adulta e 16, 7% (n = 3) apenas na adolescência (Tabela 3).
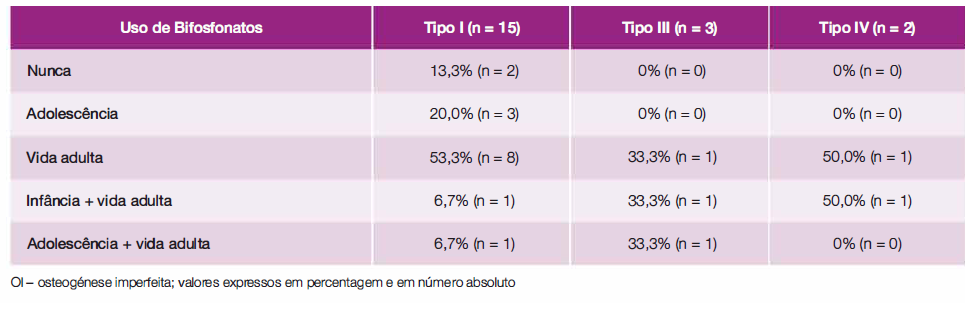
No momento em que foram avaliados os processos clínicos dos doentes, 65,0% (n = 13) dos doentes encontravam-se sob terapêutica com bifosfonatos.
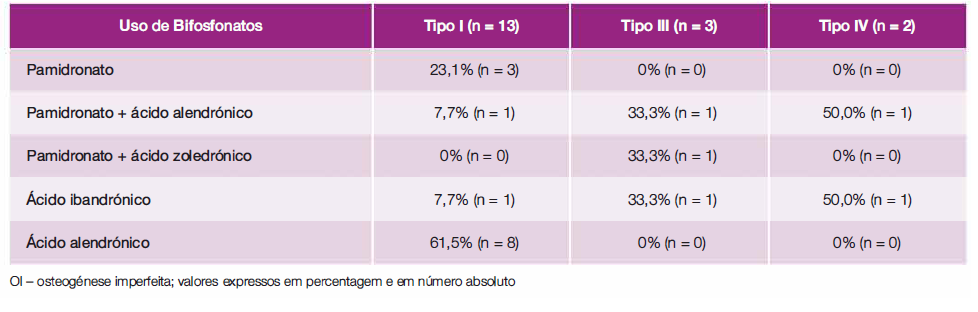
A Tabela 4 resume os bifosfonatos usados pelos doentes. O pamidronato foi o bifosfonato de escolha durante a infância e adolescência. O ácido alendrónico foi o bifosfonato mais escolhido na idade adulta. Em dois doentes do tipo I foi alterado o ácido alendrónico para ácido zoledrónico por intolerância e num doente do tipo IV foi alterado o ácido ibandrónico para ácido alendrónico pelo mesmo motivo.
Dos doentes tratados (no passado e atualmente) com bifosfonatos, foi verificada melhoria da densidade mineral óssea em seis doentes, sendo que quatro doentes não tinham densitometria óssea de seguimento e nenhum doente apresentou fraturas sob terapêutica com bifosfonatos. Dezasseis doentes cumprem suplementação de cálcio e dezoito suplementação de vitamina D. Relativamente ao tratamento não farmacológico, nove são acompanhados por fisiatria e dezassete afirmaram manter algum tipo de atividade física/reforço muscular diariamente.
Para além do seguimento em consulta de Doenças Hereditárias do Metabolismo, dois doentes mantêm seguimento por Ortopedia e doze já apresentaram necessidade de intervenção cirúrgica para correção de fraturas, através da colocação de próteses, cavilhas endomedulares ou osteossíntese com placa e parafusos. Dezoito doentes mantêm seguimento em otorrinolaringologia para avaliação periódica de défice auditivo e um doente foi submetido a estapedotomia bilateral.
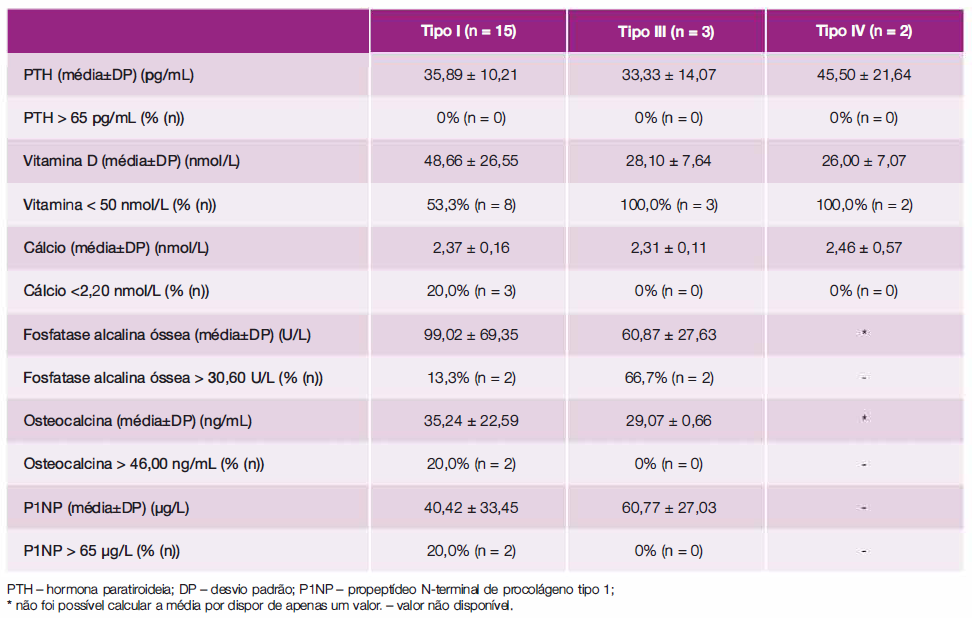
A Tabela 5 apresenta as médias dos parâmetros bioquímicos (sempre que disponíveis, previamente ao início do tratamento com bifosfonatos) de acordo com o tipo de OI, bem como a percentagem de doentes com parâmetros fora dos valores de referência. Em 65,0% (n= 13) dos doentes foram identificados baixos níveis de vitamina D, nenhum apresentou elevação da hormona paratiroideia (PTH) e a grande maioria dos doentes apresentou normocalcémia. Os marcadores de turnover ósseo, como a osteocalcina (sintetizada pelos osteoblastos) e o propeptídeo N-terminal de procolágeno tipo I (P1 NP) (marcador de reabsorção óssea) encontravam-se dentro dos parâmetros das normalidades na grande maioria dos doentes (apenas dois doentes apresentaram valores fora dos valores de referência).
Discussão
Até ao momento, esta é a primeira série de doentes adultos com OI publicada em Portugal, tendo sido já descrita a experiência do serviço de ortopedia do Hospital de Dona Estefânia, numa amostra de 21 crianças e adolescentes.7 Séries mais extensas em adultos têm sido publicadas ao longo dos últimos anos.11-15
Apesar de o diagnóstico de OI ser clínico a confirmação genética pode por vezes ser fundamental no auxílio do tipo específico. De todos os doentes com estudo genético (n = 14), 21,4% (n = 3) não tinham mutação identificada nos genes mais comuns, podendo isto ser justificado por mutações que à data do teste genético ainda não eram possíveis de detetar, pelo que podem e devem ser revisitados num futuro próximo, tendo em conta a evolução do conhecimento nesta área.
A nossa amostra apresentou um predomínio do sexo feminino para o tipo I e tipo III mas não para o tipo IV, o que está de acordo com as séries de Wekre et al12 e Scheres et al.15
A altura média dos doentes foi inferior nos doentes com tipo III o que vai ao encontro da evidência de maior deformidade neste tipo e consequente diminuição de altura.
Relativamente à OI tipo I verificou-se uma média de altura tanto para os homens como para as mulheres abaixo do valor médio descrito para a população portuguesa,16 o que traduz a baixa estatura própria destes doentes, mesmo em formas atenuadas.
A idade de diagnóstico variou com o tipo da doença tendo-se verificado que apenas 20,0% foram diagnosticados no período neonatal. Este tipo de apresentação é mais típico nos tipos mais graves (tipo III e IV) que apresentam fraturas em estadias muito precoces de vida. Dos sete doentes que foram diagnosticados em idade adulta, seis apresentavam fraturas desde infância e adolescência, o que traduz um atraso de diagnóstico desta doença que se pode prender com múltiplos fatores como a subvalorização da doença, a dificuldade no acesso aos exames complementares e ao estudo genético, bem como à própria condição social e familiar dos doentes.
Relativamente ao número de fraturas e independentemente do sexo e idade verificou-se uma média de fraturas maior nos doentes com tipo IV, seguido do tipo III, o que denota a gravidade observada nos doentes com tipo III e IV. Também nas séries descritas por Lindahl13 e Scheres,15 o tipo III apresentou maior número de fraturas face aos outros tipos de OI. No que respeita à localização das fraturas as mais frequentes foram nos ossos longos dos membros superiores e inferiores tal como é reportado na literatura. As fraturas vertebrais foram na sua grande maioria contabilizadas através da sua evidência nas radiografias do esqueleto. A grande maioria dos doentes desconhecia a existência de fraturas na coluna ainda que alguns apresentassem limitação funcional e dor crónica, amplamente desvalorizadas no contexto de dor global que a doença acarreta.
A medição da DMO através da densitometria óssea é um instrumento útil para avaliar gravidade da doença e monitorização do tratamento; quanto menor a densidade mineral óssea, maior será o risco de fratura. Ainda que na nossa amostra não tenha sido possível verificar todos os valores de Z e T score para todos os doentes tanto na coluna como no fémur proximal, verificou-se que 50,0% dos doentes (independentemente do tipo de OI) com menos de 50 anos e todos aqueles com mais de 50 anos tinham osteoporose. Este facto vem reforçar que não é mandatário para o diagnóstico que todos os doentes tenham osteoporose.17
Na nossa população os scores Z e T medidos no mesmo doente eram mais baixos na coluna lombar do que no fémur proximal na OI tipo I e tipo IV. Esta discrepância de valores em diferentes áreas do corpo é relatada também na amostra descrita por Scheres15 e a causa deste fenómeno não está claramente definida, mas pensa-se que poderá estar relacionada com a composição dos ossos em questão. Wekre12 salienta que a medição da DMO em todo o corpo seria uma forma de atenuar as limitações inerentes ao uso exclusivo de medição na coluna e fémur.
Nos doentes com OI tipo III não há registo dos valores no fémur, devido ao dano ósseo relatado previamente. Isto vai de encontro com o descrito noutras séries.15 Não se verificou diferença nos valores de DMO no fémur e coluna lombar quando comparado entre homens e mulheres, independentemente do tipo de OI, o que reforça a diferença desta doença comparativamente com a osteoporose convencional. Não houve relação entre os doentes com maior número de fraturas e aqueles com osteoporose o que mais uma vez reforça que o problema major da OI é o defeito no colagénio e não no desequilíbrio mineralização e reabsorção óssea.
Também o valor de cálcio sérico costuma ser normal nestes doentes e os valores de vitamina D baixos. Este achado, resultado de baixa exposição solar,18 é provavelmente justificada pela diminuição da mobilidade decorrente de fraturas e o receio de novas fraturas mesmo com a realização de atividades de baixo impacto, tornando estes doentes mais restritos ao domicílio.
A decisão de iniciar ou não tratamento com bifosfonato prende-se com evidência de baixa densidade mineral óssea, associada à frequência de fraturas e tolerância ao tratamento. Encontravam-se a fazer bifosfonatos 65,0% da população estudada (n = 13), sendo que comparativamente com outras séries, a nossa apresenta a percentagem mais elevada de doentes em tratamento. Apesar de treze doentes terem sido diagnosticados antes da idade adulta verificou-se que apenas sete fizeram tratamento com pamidronato, ainda que a evidência mostre maior eficácia do tratamento nas crianças.
Apenas pelos registos disponíveis não é possível apurar as causas deste facto. Ainda que não tenha sido encontrada relação entre o número de fraturas e a presença de osteoporose, na nossa amostra, houve melhoria da mineralização óssea em doentes sob tratamento com bifosfonatos e redução da incidência de fraturas o que sugere benefício em manter o tratamento nestes doentes. Assume-se que não só o tratamento farmacológico, mas igualmente o não farmacológico contribuíram para a redução da incidência de fraturas e maior estabilidade da doença.
Os doentes são monitorizados regularmente de forma a avaliar o risco de fratura e a resposta ao tratamento procurando de futuro perceber se há ou não algum tipo de tratamento que se figure melhor do que outro.
Há algumas limitações a reportar neste trabalho. A OI é uma doença rara com poucos estudos feitos na população adulta e a nossa amostra é pequena não sendo possível comparar alguns grupos com menos doentes (OI tipo III e IV) nem possível extrapolar dados para toda a população de OI adulta. Os nossos dados foram recolhidos por consulta do processo clínico e como tal algumas informações podem estar omissas. O número de fraturas e a sua localização é baseado no registo clínico dos doentes não sendo possível certificar esta informação. Em doentes com número avultado de fraturas verificou-se maior dificuldade em perceber todas as localizações, estando provavelmente este número subvalorizado.
Conclusão
A osteogénese imperfeita é uma doença com grande variabilidade clínica e com necessidade de acompanhamento de equipa multidisciplinar. A fragilidade óssea é a manifestação mais comum e mais preocupante e apesar de assistirmos a menor frequência de fraturas na idade adulta do que na infância é importante relembrar que, o risco de fratura é sustentado ao longo de toda a vida destes doentes. Ainda que menos frequentes, as manifestações sistémicas devem ser reconhecidas, uma vez que poderão ser a manifestação inaugural desta doença. Os doentes descritos estão em seguimento ativo no Centro Hospitalar sendo espectável que esta população vá aumentar nos próximos anos, não só pelos familiares dos doentes já em seguimento com caraterísticas compatíveis com OI, mas também porque o maior conhecimento da doença e das suas manifestações permite aumentar o número de diagnósticos e melhorar a sua sobrevida.
A todos os doentes em seguimento e que se encontrem em idade fértil, é dada a possibilidade de terem aconselhamento genético de forma a tentar reduzir a incidência desta patologia.

