









 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introduction
Henoch-Schönlein purpura (HSP), or immunoglobulin A vasculitis (IgAV), is the most common small-vessel vasculitis in children.1 It was formerly known as anaphylactoid purpura, although anaphylaxis is not involved in its pathophysiology.2 The disease was first described by Schönlein in 1837 as a triad of purpura, joint pain, and nephritis.3 Thirty-seven years later, his student Henoch recognized the association with gastrointestinal involvement, but it was not until 1948 that the role of immunoglobulin A (IgA) in its pathogenesis was demonstrated.4,5 Current diagnostic criteria require the presence of a palpable nonthrombocytopenic purpuric rash associated with gastrointestinal symptoms and/or arthritis and/or proteinuria/hematuria and/or histologic evidence of predominant IgA deposition.6 Central nervous system (CNS), respiratory, and genital involvement are rare. Both genetic and environmental factors have been suggested as triggers, with respiratory infections being the most important.7 The pathogenesis is not fully understood, but small vessel deposition of immune complexes containing galactose-deficient IgA is of great importance.8
The disease course is usually self-limited and only symptomatic treatment is required. In severe cases, especially in the presence of renal dysfunction, immunosuppression may be required, including corticosteroids, mycophenolate mofetil, cyclosporine, rituximab, and dapsone.8,9
In the acute phase, the outcome depends mainly on the extent of gastrointestinal involvement, while the long-term prognosis depends mainly on the extent of renal involvement. Recurrence occurs in approximately one-third of patients, but long-term follow-up is warranted in high-risk patients.2,10
Objectives
The aim of this study was to review the current evidence on the epidemiology, risk factors and pathophysiology, most common and less common clinical manifestations, diagnostic criteria, differential diagnosis, treatment options, and follow-up of IgAV.
Epidemiology and risk factors
IgAV is mostly a disease of childhood, with 90% of affected individuals under the age of 10 and a mean age of diagnosis of six years.2,9 The incidence is probably underestimated at 10-56 children per 100,000 per year due to underreporting.11,12 There is a predominance of cases in winter.1,9 Several risk factors for renal involvement have been described, and the most important contributors include: older age of onset (especially over 10 years) and wintertime, longer interval between symptom onset and diagnosis, rural residence, persistent purpura, severe gastrointestinal symptoms, recurrence, angioedema, obesity, and decreased C3.1,10,13 Data on sex predominance are conflicting, but most studies suggest a slight male predominance of 1.2-1.8:1.11 The role of sex in renal manifestations is uncertain.11) IgAV is reported worldwide, but is less observed in subjects of African descent.2,8
In most cases, the precipitating cause is not identified, but there is often an association with preceding infections, usually in the upper respiratory tract.7,14 A correlation between virtually all respiratory pathogens and IgAV has been observed.9 Associated pathogens are listed in Table 1 and include Streptococcus, particularly Group A β-hemolytic Streptococcus, parvovirus B19, hepatitis B virus, hepatitis C virus, human immunodeficiency virus 1, Bartonella henselae, Salmonella spp, Shigella, Staphylococcus aureus, and Mycoplasma pneumoniae.9) Less common viral pathogens include Epstein-Barr virus, adenovirus, coxsackie virus, hepatitis A, varicella zoster virus, citomegalovirus, influenza, rotavirus, norovirus, and bocavirus.2,8,14-16 Besides infections, also foods, immunizations, drugs, and allergens can trigger IgAV.1 Drug-induced cases are more common in adults, and angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor antagonists (losartan), antibiotics (clarithromycin), L-dopa, chlorpromazine, and nonsteroidal anti-inflammatory drugs (NSAIDs) have been implicated.2,14 Cases of IgAV have been reported following severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection in children, and one case following coronavirus disease-19 vaccination in an adult.17-19 At least five cases have been reported following hymenoptera sting.20
Table 1 Most common pathogens associated with IgAV
| Bacteria |
| Streptococcus (group A β-hemolytic Streptococcus) |
| Staphylococcus aureus |
| Bartonella henselae |
| Salmonella spp. |
| Shigella spp. |
| Mycoplasma pneumoniae. |
| Viruses |
| Adenovirus |
| Bocavirus |
| Cytomegalovirus |
| Coxsackie virus |
| Epstein-Barr virus |
| Hepatitis A virus, hepatitis B virus, hepatitis C virus |
| Human immunodeficiency virus 1 |
| Influenza virus |
| Norovirus |
| Parvovirus B19 |
| Rotavirus |
| Varicella zoster virus |
Pathophysiology
IgAV is a non-granulomatous leukocytoclastic vasculitis that affects small vessels.14 Leukocytoclasia originates from partially disintegrated neutrophil granulocytes in the vessel walls that are deposited around the extravasated erythrocytes that cause purpura.9
Although the detailed pathogenic mechanisms of IgAV are not fully understood, IgA plays an important role. IgA deposition has been found in patients with IgAV, IgA nephritis (IgAN), skin-limited IgA, and systemic IgA, suggesting a common pathophysiology.2,8 An inherited defect is likely the initial trigger for the glycosylation abnormalities that occur in the O-linked oligosaccharides of the IgA1 hinge region, particularly in galactose and sialic acid levels.14,21 The abnormal IgA1 is inappropriately catabolized in the liver, leading to its accumulation and deposition. In addition, IgA1 is particularly polymeric, facilitating the formation of immune complexes with unaltered IgA1 and IgG.21 Environmental triggers such as infections are thought to stimulate IgA production.8 Both quantitative and structural abnormalities in IgA cause overexpression of RFcI (CD89), found on circulating monocytes and neutrophils, and the transferrin receptor (CD71), found on mesangial cells. IgA also forms immune complexes with CD89, which then bind to CD71 and are deposited in the walls of small vessels.22 Once deposited, these immune complexes activate the alternative complement pathway, causing C3 deposition and further damaging the endothelial lining by promoting mesangial cell proliferation and the local inflammation in the glomeruli responsible for progressive renal failure.7,8 There are several molecules involved in local inflammation that are elevated in the acute phase of IgAV, originating from both immune and non-immune cells.9) These include antibodies, receptors, transmembrane proteins, endothelins (ET-1), vasoconstrictor hormones and cytokines such as interleukin (IL)-1, IL-6, and IL-8, toll-like receptors (TLR)-2 and TLR-4, and tumor necrosis factor-α (TNF-α), which also induces adhesion molecules on endothelial cells and leukocytes.2,9,13,21 The roles of IgG and IgM in IgAV are still being elucidated.21
The familial association in the occurrence of IgAV suggests the important role of genetic contribution, particularly mutations in the Mediterranean fever gene, which encodes the protein pyrin/marenostrin, which then regulates caspase-1 activation and IL-1B production. There is an increased risk for IgAV associated with human leukocyte antigens (HLA) A2, A11, B35, B34, and DRB1*01, and a decreased risk in carriers of HLA-A1, DRB1*07, B49, and B50.2,9, 14(,(16 HLA-B35 is particularly associated with renal involvement.2
There is recent evidence regarding the role of the digestive system in the pathophysiology of IgAV, such as the involvement of gliadin in mouse models and improvement with gluten-free diets, the activation of the innate immune response in the digestive mucosa, the association of IgAN with genes involved in immunity to intestinal microorganisms and in inflammatory bowel disease, the particular composition of the microbiota in these patients, and the improvement with ileum-targeted release formulations of budesonide.8
Clinical manifestations
The classic tetrad of IgAV consists of palpable purpura, arthralgia (joint pain), gastrointestinal complaints, and renal involvement. Skin involvement is mandatory. The order of presentation varies, but the clinical course most commonly begins with purpura and joint pain and progresses over days to weeks. In most cases, the diagnosis is made in less than four days.1 Rarely, the CNS, testis, and lungs are also affected.9 Less common manifestations include low-grade fever, malaise, orchitis, seizures, epistaxis, ileoileal intussusception, jejunal hemorrhage, orbital hematoma, CNS bleeding, and ischemic stroke.2
Skin
The rash usually begins with petechiae and palpable purpura. Non-blanching, round, oval, and retiform indurations up to 1 cm in diameter appear predominantly on the lower limbs and extensor surfaces, usually in a symmetrical distribution (Figure 1).8,14,23 Slower blood flow in the dilated venules favors deposition of IgA complexes on the lower limbs and extensor surfaces, but lesions also occur on the forearms, cheeks, and ears in one-third of patients.9 Rarely, early lesions may be erythematous maculae or urticarial, which resolve with pressure. Bullous or necrotic lesions are less common, especially in children.2,9) Lesions resolve completely over time, but affected areas remain discolored for weeks after resolution due to hemosiderin deposition.9 Non-pitting edema and pruritus may also be present.2
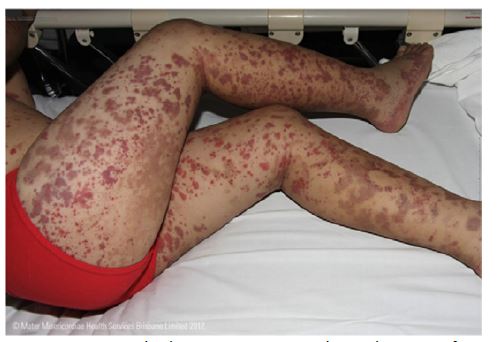
Joints
Arthralgia or arthritis affects 75% of children with IgAV and precedes the onset of purpura in up to 25% of cases and up to 14 days. The joints become painfully swollen, with limited mobility, but spontaneous resolution occurs without joint destruction.2,9 One or more joints may be affected, either simultaneously or not. Knees and ankles are most commonly affected, but hands and feet can also be affected.8
Gastrointestinal tract
In up to 40% of patients, the clinical course begins with gastrointestinal manifestations. Deposition of immune complexes in the intestinal vessel walls leads to abdominal pain, vomiting, melena, and hematemesis. Abdominal pain, usually epigastric or periumbilical, is the most common symptom.2 Hypoalbuminemia without proteinuria, fecal calprotectin measurement, fecal occult blood testing, and imaging studies may be useful in detecting subclinical intestinal involvement.2,8,9 Upper and lower digestive endoscopic study allows direct analysis and biopsy of lesions, which are more commonly found in the duodenum and may present as mucosal erythema, petechial purpura, areas of necrosis, or erosion.8 In severe cases, differential diagnosis with acute surgical abdomen is necessary. Although rare, complications are more common in children than in adults and include perforation, intussusception, bowel angina, and infarction. Because of the potential severity, acute mortality associated with IgAV is mostly related to gastrointestinal involvement.7,9,14
Kidney (Henoch-Schönlein purpura nephritis)
IgAV nephritis (IgAVN) affects 20-80% of IgAV patients, usually within one to three months of IgAV onset, but may also occur several months later, with or without purpura relapse.1,2,8,11 It is the most important source of morbidity associated with IgAV. Microscopic hematuria and mild proteinuria are the most common and earliest findings.24 Nephrotic and nephritic syndrome occurs in 7% of patients and end-stage renal disease in 1-2%, possibly more than ten years after onset.8 If renal involvement is absent within six months, long-term renal damage is highly unlikely.9 Renal failure at diagnosis is extremely rare in children. Hypertension may develop at any time during the course of the disease.11
Onset over six years of age and during colder seasons, time from initial manifestations to diagnosis over eight days, persistent purpura over one month, rural residence, recurrence, angioedema, gastrointestinal bleeding, and CNS involvement are significant risk factors for renal involvement.1,2 The course is usually self-limited and benign. However, the degree of proteinuria is the most important determinant of IgAV prognosis.1 Age, platelet distribution width, higher CD3, lower fibrinogen, C-reactive protein, and neutrophil-to-lymphocyte ratio, and higher IgG may be useful in predicting renal involvement in IgAV patients.11 Pathologically, it is determined by the presence of crescents or tubulointerstitial changes.2 IgA levels do not seem to correlate with renal involvement. This is an important cause of renal failure in children, accounting for 3% of renal replacement therapy cases in pediatrics.8
Central nervous system
This vasculitis affects CNS tissues by causing edema, ischemia, infarction, thrombosis, and/or hemorrhage.8,14 It usually occurs two to four weeks after the onset of HSP, mostly in patients with severe disease and concomitant multi-organ involvement.14,25 The posterior parietal and occipital regions are more commonly involved, possibly due to facilitated IgA deposition in the posterior circulation.14 Headache is the most common symptom, followed by seizures, irritability, dizziness, emotional instability, behavioral changes, hemiparesis and other focal deficits, nystagmus, ataxia, aphasia, and dysarthria. Rare complications include intracranial hemorrhage, mononeuropathy, chorea, acute motor sensory axonal neuropathy, and posterior reversible encephalopathy syndrome.9,26 Despite its low prevalence, 20% of cases result in permanent sequelae, such as visual and verbal deficits, focal signs, and epilepsy.14
Magnetic resonance imaging may show cerebral vasculitis.8 Lumbar puncture is usually normal but can be relevant in the differential diagnosis.14
Lungs
Although rare (0.8-5% of IgAV cases), pulmonary involvement is often severe, with a mortality rate of 27%, mostly due to diffuse alveolar hemorrhage, which is the most common presentation.8 Deposition of immune complexes in the alveolar-capillary membrane causes pulmonary edema, intra-alveolar hemorrhage, interstitial fibrosis, arterial neutrophilic infiltrate, loss of nuclear staining in the alveolar walls and septa, and capillary wall necrosis.27 Most patients also present with renal involvement, and antineutrophilic cytoplasmic antibody (ANCA) vasculitis should be considered in these cases.8 Presentation ranges from cough, hemoptysis, dyspnea, tachypnea, and chest pain to acute respiratory failure. Occasionally, subclinical changes are noted on pulmonary function test or chest radiograph. Lung biopsy confirms leukocytoclastic vasculitis. First-line treatment is high-dose intravenous pulse methylprednisolone with or without immunosuppressive therapy.27
Other rare complications include parotitis, myositis, episcleritis, carditis, myocarditis, pancreatitis, cholecystitis, pseudomembranous colitis, orchitis/epididymo-orchitis, and urethritis.8,28-30
Diagnosis
The diagnosis of IgAV is based on clinical manifestations and histologic findings, as there are currently no proven useful biomarkers.7,9 In 2010, the European League Against Rheumatism (EULAR), the Paediatric Rheumatology International Trials Organization (PRINTO), and the Paediatric Rheumatology European Society (PRES) published revised criteria for the diagnosis of IgAV in children, achieving 100% sensitivity and 87% specificity.9 Predominant purpura or petechiae in the lower limbs is a mandatory criterion, plus at least one of the following: acute-onset diffuse abdominal pain; acute-onset arthritis or arthralgia; renal involvement (proteinuria or hematuria); leukocytoclastic vasculitis or proliferative glomerulonephritis with predominant IgA deposition on histopathology.6
As with any cutaneous vasculitis, skin biopsy is the gold standard for the diagnosis of IgAV. IgA-predominant vascular deposits are the most characteristic IgAV finding, but are not always present and may be caused by other conditions such as vasculitic syndromes, erythema nodosum, and venous stasis.9 Direct immunofluorescence may also show perivascular infiltration of neutrophils and mononuclear cells and C3, IgG (40% of cases), and IgM (20% of cases) deposits, reflecting the presence of immune complexes.2,8,14 Skin biopsy is usually recommended only when clinical criteria are not met.9 Histologic evidence of leukocytoclastic vasculitis includes inflammation and fibrinoid necrosis of the vessel walls, endothelial swelling, neutrophilic infiltrate with nuclear fragmentation, and deposition of cellular debris in the surrounding skin (Figure 2).2
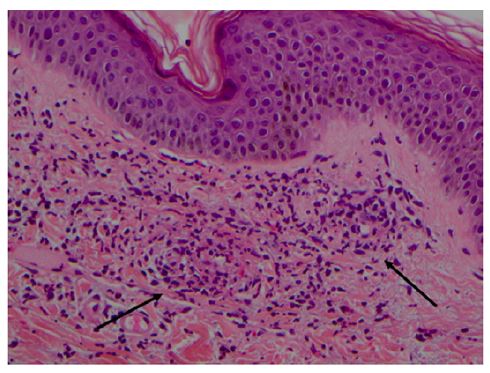
Figure 2 Leukocytoclastic vasculitis of the skin in a child with Henoch- Schönlein purpura. Superficial dermal vessels with inflammatory infiltrate composed mainly of neutrophils and eosinophils (arrows). Hematoxylin/eosin; magnification × 200 (courtesy of the author of Reference 42).
Kidney biopsy is recommended in the presence of persistent severe proteinuria, but may also be considered in cases of short-term severe proteinuria, persistent moderate proteinuria, or impaired glomerular filtration rate.31 It may show a wide variety of glomerular lesions, most commonly focal and segmental glomerulonephritis.14 Mesangial deposits in sclerotic glomeruli allow for retrospective diagnosis.8
The current histologic classification for IgAVN in children considers only mesangial proliferation and crescent ratio. Therefore, some authors advocate the use of the MEST-C score according to the 2016 revision of the Oxford classification for IgAN, which considers mesangial hypercellularity (M), endocapillary hypercellularity (E), segmental glomerulosclerosis (S), tubular atrophy/interstitial fibrosis (T), and cellular/fibrocellular crescents. However, this classification awaits validation in pediatrics and IgAVN.32
Because biopsies are not always easily obtained and many vasculitides and glomerulonephritides share similar histopathologic findings, the development of less invasive, disease-specific, and early-detectable biomarkers is of great interest.7,11 Laboratory tests can aid in differential diagnosis and early detection of complications.14 A complete blood count may show moderate leukocytosis and normal or elevated platelet counts; coagulation studies are normal.2,7 Acute phase reactants may be slightly elevated. Antinuclear antibodies are usually undetectable.7) Serum C3 and C4 levels may be decreased, especially in children with a recent streptococcal infection.33 Half of patients show an increase in serum IgA, but it is unclear whether these are clinically significant.2,21 Regarding renal involvement, urinalysis should be performed at the time of diagnosis, and sequential assessments are recommended three to six months after IgAV onset. It may reveal proteinuria, and the presence of erythrocyte casts or dysmorphic red blood cells indicates the glomerular origin of hematuria.
The differential diagnosis includes acute hemorrhagic edema of infancy (AHEI), leukemia and leukemic lymphoma, acute post-streptococcal glomerulonephritis (APSGN), septicemia, disseminated intravascular coagulation, papular-purpuric gloves-and-socks syndrome, Mediterranean fever, immune thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and other vasculitides such as hypersensitivity, urticarial and ANCA-associated small vessel vasculitis, mixed cryoglobulinemia, cutaneous polyarteritis, rheumatic diseases such as systemic lupus erythematosus, rheumatoid arthritis, Sjögren’s syndrome, mixed connective tissue disease, juvenile dermatomyositis, or antiphospholipid antibody syndrome. The gastrointestinal presentation may be confused with causes of acute surgical abdomen.2,7
In AHEI, purpuric lesions involve the lower extremities as in IgAV, but also the face and are usually larger, sometimes target-shaped or developing into bullae, and preceded by facial edema. Peripheral edema is also common, and gastrointestinal or renal involvement is rare. Vascular IgA deposits are rarely seen.2
The signs of leukocytoclastic vasculitis may also be found in cases of leukemia and leukemic lymphoma.2 Skin biopsy may be useful in differentiating from other vasculitides.7 Vasculitis may be a manifestation of malignancy, either hematologic or solid tumors of gastrointestinal, respiratory, or urinary origin.9 In the presence of hematuria, renal biopsy helps to differentiate from APSGN by showing diffuse granular IgG deposits.2
In the future, the study of IgA1 binding sites and other antibodies such as IgA rheumatoid factor, IgA anticardiolipin antibodies, and IgA anti endothelial cell antibodies may be relevant to the diagnosis.7 IgA anti-beta2 glycoprotein antibodies are elevated in IgAV, especially in association with severe renal and joint involvement.9
Treatment/Management
In the absence of renal involvement, supportive treatment is the backbone of this mostly benign and self-limited disease, consisting of hydration, electrolyte balance, and nutrition.2,7 Compression therapy and antihistamines can help reduce vasodilation and increase blood flow, thereby inhibiting immunoglobulin deposition and lesion progression. Immobilization should be limited to those with severe joint involvement, as it increases the risk of thrombosis, does not affect gastrointestinal or renal involvement, and is beneficial in containing purpuric lesions.8 Analgesics and antispasmodics are preferred to NSAIDs because of the hemorrhagic and nephrotoxic effects of the latter.8
The use of corticosteroids is controversial due to side effects such as weight gain, moon facies, and generalized edema, but may be useful in cases of painful edema and incipient necrosis prior to ulcer formation.2
The optimal approach to IgAVN remains uncertain and is often extrapolated from IgAN, especially in cases of more severe renal involvement.8,9 These patients may require renin-angiotensin system blockers (ACE inhibitors or angiotensin II receptor antagonists) to control blood pressure and persistent proteinuria.8 If proteinuria remains above 1 g/day/1.73m2, methylprednisolone pulses followed by a short course of oral corticosteroids are usually recommended.34 The use of terminal ileum-targeted budesonide shows promise in providing effective results with fewer side effects.35 Corticosteroids appear to have no effect in preventing renal damage, but have a potential benefit in improving the course of established renal involvement, significantly reducing proteinuria and subsequent end-stage renal disease, particularly if started before fibrous crescents are found.2,36 They may be used independently or in combination with cytotoxic agents, particularly in rapidly progressive glomerulonephritis.2 There are conflicting data regarding the benefit of cytotoxic agents. Some studies have found no benefit from the use of cyclophosphamide alone or in combination with corticosteroids, while others have demonstrated the advantage of cyclophosphamide and azathioprine in inducing remission and improving its duration.2,9,37 Azathioprine is usually preferred to cyclophosphamide because of its lower toxicity.2 Mycophenolate mofetil has also shown better results than cyclophosphamide in reducing proteinuria, with fewer side effects.9,38 Rituximab, an anti-CD20 antibody, inhibits B-cells and has been effective in inducing remission in individual cases where it has been used, even in end-stage renal disease.9
Compared to methylprednisolone, cyclosporine A showed no difference in biopsies at two years, but more frequent and faster regression of proteinuria.9 It may be used in corticosteroid-dependent patients.2 Anticoagulants have also been studied for their potential role in limiting fibrin accumulation, but are generally not recommended due to the risk of adverse effects.2,9,37 O-aminocaproic acid, methotrexate, and hydroxychloroquine have been studied in adults with IgAV.2,8 Ongoing studies are evaluating the contribution of colchicine in skin-limited vasculitis.39 The prophylactic role of tonsillectomy has been discussed but is not usually recommended.9 The use of immunoglobulins and plasmapheresis reduces circulating immune complexes and appears to be beneficial in combination with other therapies, but more studies are needed to clarify their role.2,8,9
Glucocorticoids may be helpful for abdominal and joint pain after symptomatic measures have failed.2,8 In particularly severe cases and after corticosteroid failure, further immunosuppressive strategies may be considered.38 Abdominal surgery may be necessary for gastrointestinal complications.9 After exclusion of glucose-6-phosphate deficiency, dapsone has been used in specific cases of persistent and severe purpuric lesions, with complete resolution.8,9
Prognosis and follow-up
The acute phase of IgAV usually lasts one to four weeks.2 Although it is mostly a benign disease, 12% of children develop chronic kidney disease within three to four years, and 20% of these progress to end-stage renal disease.2 Follow-up and close monitoring are recommended until complete clinical and laboratory resolution.8 Thereafter, the risk of renal impairment decreases dramatically, and there is no consensus regarding follow-up, but annual observations have been advocated.8,40 Children with renal involvement should be followed long-term.12 Factors associated with poor prognosis include nephrotic and nephritic syndrome, acute kidney injury at presentation, hypertension, proteinuria/creatinuria ratio greater than 1 g/g, and more than 50% crescent or interstitial fibrosis on renal biopsy.2,8 In contrast, children who do not present with urinary abnormalities within the first six months rarely have a decline in renal function.8 Recurrence is defined by the recurrence of symptoms at least two weeks after resolution, but usually occurs within three to four months and affects approximately 15-40% of patients.2 The most common manifestations are gastrointestinal and cutaneous. Predicting recurrence is challenging as there are currently no validated biomarkers with prognostic value and the association with older age is controversial.8,41 Low-dose corticosteroids in alternate-day regimens and dapsone may have a role in preventing relapse.2
Conclusions
IgAV primarily affects children, often following a respiratory infection as trigger.7 In the absence of renal involvement, supportive treatment is sufficient.7 Corticosteroids may be useful in cases of severe gastrointestinal manifestations, but do not alter the incidence of nephritis and are therefore not recommended for the prevention of kidney disease.2 Despite its usually benign course in children, IgAV has the potential to induce chronic kidney disease and even end-stage renal disease.8 The extent of renal involvement greatly influences the long-term prognosis.10 As new evidence emerges, a better understanding of the physiopathology underlying IgAV is gained and better therapeutic options arise. However, there is still an important lack of early and non-invasive diagnostic and prognostic biomarkers to guide the optimal management of these patients.11
Authorship
Patrícia Sousa - Conceptualization; Data curation; Formal Analysis; Investigation; Methodology, Writing - original draft
Susana Correia de Oliveira - Data curation; Formal Analysis; Investigation; Writing - review & editing
Angela Dias - Conceptualization; Investigatio; Methodology; Writing - review & editing
Cláudia Tavares - Conceptualization; Project administration; Supervision; Writing - review & editing