










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkNascer e Crescer
versão impressa ISSN 0872-0754versão On-line ISSN 2183-9417
Nascer e Crescer vol.29 no.2 Porto jun. 2020
https://doi.org/10.25753/BirthGrowthMJ.v29.i2.15184
CASE REPORTS | CASOS CLÍNICOS
Coarse face, hypotonia, and neurodevelopmental regression
Face grosseira, hipotonia e regressão do neurodesenvolvimento
Ana Margalha MirandaI, Marta EzequielI, Catarina LuísI, Juliette DupontII, Paulo GasparIII, Laura VilarinhoIII, Patrícia JaneiroIV, Ana GasparIV
I. Department of Pediatrics, Hospital Prof. Doutor Fernando Fonseca. 2720-276 Amadora, Portugal. ana.margalha.miranda@gmail.com; marta.ezequiel@gmail.com; csmrluis@gmail.com
II. Department of Genetic, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte. 1649-035 Lisboa, Portugal. jdupontg@gmail.com
III. Metabolism and Genetic Unit, Department of Human Genetic, Instituto Nacional de Saúde Dr. Ricardo Jorge. 4000-053 Porto, Portugal. paulo.gaspar@insa.min-saude.pt; laura.vilarinho@insa.min-saude.pt
IV. Reference Centre of Inherited Metabolic Diseases, Department of Pediatrics, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte. 1649-035 Lisboa, Portugal. patricia.janeiro@gmail.com; ana.m.gaspar@chln.min-saude.pt
Endereço para correspondência | Dirección para correspondencia | Correspondence
ABSTRACT
Inborn errors of metabolism are a heterogeneous class of multisystemic diseases which, although individually rare, are collectively quite common. Central nervous system is usually affected.
The authors report the case of a five-month-old girl, daughter of non-consanguineous parents, born after an unremarkable full-term pregnancy and delivery. Hypotonia and neurodevelopmental regression were noted from the age of five months, along with progressive onset of facial dysmorphism, hepatomegaly, seizures, and dilated cardiomyopathy. Gangliosidosis type 1 diagnosis was confirmed by biochemical, enzymatic, and genetic findings.
This report enhances the relevance of multidisciplinary approach and follow-up.
Keywords: Coarse facies; developmental regression; Gangliosidosis; hypotonia; lysosomal storage disease
RESUMO
Os erros hereditários do metabolismo são um grupo de doenças heterogéneas e multissistémicas. Apesar de cada doença individualmente ser rara, no seu conjunto são relativamente comuns. O sistema nervoso central é habitualmente afetado.
Os autores apresentam o caso de uma lactente de cinco meses de idade, filha de pais não consanguíneos, nascida de uma gravidez de termo e parto sem intercorrências. Aos cinco meses, iniciou um quadro de hipotonia e regressão do desenvolvimento, apresentando ao longo do tempo dismorfismo facial, hepatomegália, convulsões e cardiomiopatia dilatada. O diagnóstico de Gangliosidose tipo 1 foi confirmado por achados bioquímicos, enzimáticos e genéticos.
Este caso clínico reforça a relevância de uma abordagem e seguimento multidisciplinares.
Palavras-chave: Doença lissosomal de sobrecarga; face grosseira; Gangliosidose; hipotonia; regressão do desenvolvimento
Introduction
Inborn errors of metabolism (IEMs) are a wide and heterogeneous group of diseases resulting from the absence or deficiency of an enzyme or its cofactor, leading to either accumulation or deficiency of specific metabolites. Each hereditary metabolic disease is a rare entity, but collectively they represent a frequent clinical finding. IEMs can affect any organ or system. Neurological are among the most common findings, namely developmental delay or regression, epilepsy, or abnormal muscular tone. Dysmorphic features can also occur in some diseases in this group and may be present from birth or develop throughout life.1
Lysosomal storage disorders (LSD) are one group of IEMs in which the enzymatic defect leads to accumulation of undegraded insoluble metabolites within lysosomes. Gangliosidosis type 1 (GM1) is a severe LSD with important central nervous system (CNS) involvement.1
Case report
The authors report the case of a Caucasian girl referred at the age of five months to the General Pediatric consultation due to recurrent wheezing.
The girl was the second child of healthy non-consanguineous parents. Her ten-year-old brother was healthy. No family history of genetic, neurological, or metabolic diseases and no previous sibling or fetal deaths were acknowledged.
Pregnancy was uneventful, with normal prenatal ultrasounds. The girl was born at 37 gestational weeks and six days by spontaneous vaginal delivery, with growth restriction: she weighed 2410 g (3−10th percentile for gestational age and sex - Fenton growth chart2), was 45 cm height (3−10th percentile for gestational age and sex - Fenton growth chart), and had 32.5 cm of head circumference (10−50th percentile for gestational age and sex - Fenton growth chart).2 No complications were reported during delivery or perinatal period. Neonatal hearing screening with acoustic otoemissions revealed bilateral absence of evoked otoacoustic emission response, suggesting congenital hearing loss. Evoked audition potentials were inconclusive, but the exam could not be repeated. Congenital heart disease screening (with pulse oximetry) was normal, as well as neonatal metabolic screening. The patient had two previous admissions due to respiratory infection, at 3 and 4.5 months, with recurrent wheezing as sequelae. After discharge, she received inhaled fluticasone and was referred to the General Pediatric consultation.
Physical examination at five months showed non-suspicious facial features. Cardiopulmonary auscultation and abdominal evaluation were normal, with no cardiac murmur or organomegaly. Neurological exam revealed predominantly bilateral fisted hand with forearm in a flexed and supine position. The patient held the head in seated position, maintained eye contact, and smiled. The remaining physical observation was normal.
At the age of six months, a significant neurodevelopmental regression was noted, with severe hypotonia and poor cephalic control. Hepatomegaly was also noted, but no facial abnormalities.
Ongoing follow-up revealed progressive facial feature change (Figure 1). Mild frontal bossing, thick eyebrows, depressed nasal bridge, and long philtrum were evidenced, as well as macroglossia, gingival hypertrophy, thickened skin, hirsutism, dorsal kyphosis, flexion hand contractures, and prominent abdomen with progressive hepatomegaly. At nine months, the girl was diagnosed with dilated cardiomyopathy (end-diastolic diameter of the left ventricle: 46 mm - Z-score 16) with left ventricular systolic function at the lower limit of normal (shortening fraction 28%) and moderate-to-severe mitral regurgitation due to anterior leaflet prolapse and annulus dilatation. At 11 months, she had her first seizure with generalized tonic-clonic movements.
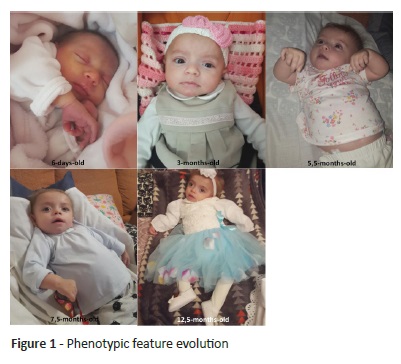
Although neuromuscular and skeletal symptoms progressed, the patient started physiotherapy at the age of six months. Weight and height evolved on the 15th percentile and head circumference on the 15-50th percentile for age and sex, according to WHO Growth Charts.3
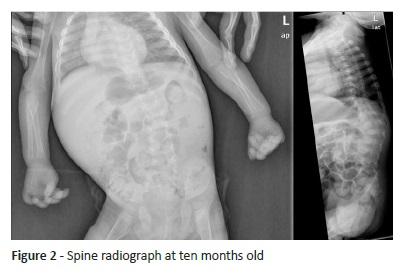
Developmental regression preceded dysmorphic findings, leading to an extensive study. Magnetic resonance imaging, performed at 6.5 months, showed narrowed corpus callosum thickness, slight normal myelinisation delay of cortical-spinal tract, and normal ventricle, brain, and cerebellum dimension. Ophthalmological examination revealed divergent strabismus with normal fundoscopy. Spine x-ray revealed severe scoliosis with anterior angle in dorsal and lumbar spine transition (Figure 2). Electroencephalography (at 11 months) showed diffuse lentification, with no seizure focus.
Initial metabolic evaluation including ammonia, free amino acid and organic acid chromatographic analysis, redox state, and very long-chain fatty acids was normal.
Lysosomal enzymatic activity measured in dried blood spots was low for α-L-iduronidase (0.08 nmol/h/spot; NR 0.10-0.9) and β-galactosidase (0.05 nmol/h/spot; NR 1.50-7.00) activity. The remaining enzymatic activity tests (iduronate-2-sulfatase, N-acetil-α-D-glucosaminidase, galactosamine-6-sulfatase, arylsulfatase B, and β-glucuronidase) were all normal. Low β-galactosidase activity suggested Gangliosidosis type 1 (GM1) or Morquio disease type B.4 Glycosaminoglycans measured in urine were increased (104 mg/mmol creatinine; normal range 4−31) and monodimensional electrophoresis revealed chondroitin-6-sulphate accumulation. Absence of increased keratane sulphate urinary excretion, along with clinical storage in neural tissues indicated GM1 as the most likely diagnosis.
Definite GM1 diagnosis was established following identification of a compound heterozygous mutation (c.588-591insT and c.1581G>A) on GLB1 gene at the age of 11 months.4 The girl’s parents were heterozygous for one of the mutations.
The girl was hospitalized several times due to pulmonary infections. She presented generalized hypotonia, lethargy, occasional seizures (partially controlled with levetiracetam and phenytoin) and profound mental retardation. Feeding skills persisted until she was 16 months old. After receiving palliative support, the girl died at the age of 18 months. The family currently benefits from psychological support.
Discussion/conclusion
GM1 is an autosomal recessive LSD caused by β-galactosidase enzyme deficiency.4,5 It is a rare disease with an estimated incidence of 1 in 100,000-200,000 live births.4
GM1 can be classified into three types based on clinical phenotype, with disease severity inversely correlated with residual activity of the mutant enzyme.4 Type 1 or infantile form is the most severe form of disease, with onset between birth and six months, rapidly progressive course, and death by the age of 1-2 years.4 Classical symptoms are present in almost every patient by the time of diagnosis: coarse facial features (87%), cherry-red macula (59%), hepatosplenomegaly (85%), and skeletal dysostosis (82%). Cardiomyopathy is present in one third of GM1 cases and CNS involvement in all cases.4
β-galactosidase enzyme is encoded by GLB1 gene (E.C.3.2.1.23; MIM 230500), mapping on the 3p21.33 chromosome with 16 exons.6 More than 130 genetic alterations have been reported in GLB1 gene to date. According to the literature, many patients are compound heterozygous, as in the present case, which confirms the extensive molecular heterogeneity of β-galactosidase gene and the difficulty in correlating genotype with phenotype.6,7
Enzymatic deficiency leads to a massive storage of GM1 ganglioside and related glycoconjugates (oligosaccharidosis and mucopolysaccharidosis) in different tissues, namely CNS, with progressive tissue destruction characteristic of GM1.4,8 Extraneuronal clinical involvement is related to oligo and mucopolysaccharide accumulation. Some authors suggest that phenotypic variability is explained by different substrate binding sites, protein folding-relevant sites, or enzyme subdomains, required for catabolism of different substrates. Consequently, β-galactosidade mutations produce phenotypes with different degrees of neuronal dysfunction and extraneuronal signs and symptoms.4,8
GLB1 gene also encodes for elastin binding protein (EBP), located in the endosomal compartment. Its depletion in arterial smooth muscle, fibroblasts, and chondroblasts interferes with elastic fibre assembly, probably leading to skeletal and cardiac deformations.9,10 Santamaria et al. reported that all cardiac disease patients had two GLB1 mutations affecting both lysosomal enzyme and EBP, suggesting that cardiac involvement is caused by an homozygous EBP alteration.6 However, findings from this study argue against this, as the patient only had one EBP-affecting mutation (c.1581G>A). Some authors argue that elastogenesis can also be disrupted by pericellular accumulation of galactosugar-bearing-moieties, such as chondroitin sulphate or dermatan sulphate, which bind to EBP and induce both its shedding from cell surface and premature tropoelastin release far from microfibrillar acceptors.10 This second mechanism acts as a secondary EBP defect and could help understand cardiac disease in the present patient.
There is no curative GM1 treatment at present, but therapeutic options are being investigated.
Until curative treatment emerges, all children and their families should be referred to palliative care for end-of-life support and quality of life improvement.4
In conclusion, GM1 is a rare disease with multisystemic involvement. Early diagnosis is challenging due to clinical heterogeneity, sign and symptom similarity with other IEMs, and their progressive onset over time. Awareness of multisystemic and progressive involvement may be key to reach an early diagnosis and offer a complete and multidisciplinary therapeutic approach, together with genetic counselling including prenatal diagnostic options for the family.
Consent
Informed consent for case, image, and laboratory study publication was previously obtained from the patient’s parents. Parents authorized case publication without feature hiding.
REFERENCES
1. Sutton VR. Inborn errors of metabolism: Epidemiology, pathogenesis, and clinical features. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. http://www.uptodate.com (Accessed on May 17, 2018). [ Links ]
2. Fenton TR, Kim JH. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatrics. 2013; 13:59. [ Links ]
3. The WHO Child Growth Standards. 2007. (Assessed May 28, 2020). Available at: https://www.who.int/childgrowth/standards/en/ [ Links ]
4. Brunetti-Pierri N, Scaglia F. GM1 gangliosidosis: Review of clinical, molecular, and therapeutic aspects. Molecular Genetics and Metabolism. 2008; 94:391-6. [ Links ]
5. Erol I, Alehan F, Pourbagher MA, Cana O, Vefa Yildirim S. Neuroimaging findings in infantile GM1 gangliosidosis. European Journal of Paediatric Neurology. 2006; 10:245-8. [ Links ]
6. Santamaria R, Chabás A, Coll MJ, Miranda CS, Vilageliu L, Grinberg D. Twenty-one Novel Mutations in the GLB1 Gene Identified in a Large Group of GM1-Gangliosidosisand Morquio B Patients: Possible Common Origin for the Prevalent p.R59H Mutation Among Gypsies. Hum Mutat. 2006; 27:1060. [ Links ]
7. Silva C, Severini M, Sopelsa A. Six Novel β-Galactosidase Gene Mutations in Brazilian Patients With GM1-Gangliosidosis. Human Mutat. 1999; 13:401-9. [ Links ]
8. Sandhoff K, Harzer K. Gangliosides and Gangliosidoses: Principles of Molecular and Metabolic Pathogenesis. The Journal of Neuroscience. 2013; 33:10195-208. [ Links ]
9. Caciotti A, Garman SC, Rivera-Colón Y. GM1 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochim Biophys Acta. 2011; 1812:782-90. [ Links ]
10. Hinek A, Zhang S, Smith AC, Callahan JW.Impaired Elastic-Fiber Assembly by Fibroblasts from Patients with Either Morquio B Disease or Infantile GM1-Gangliosidosis Is Linked to Deficiency in the 67-kD Spliced Variant of b-Galactosidase Am J Hum. Genet. 2000; 67:23-36. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Ana Margalha Miranda
Department of Pediatrics
Hospital Prof. Doutor Fernando Fonseca.
IC19, 2720-276 Amadora
Email: ana.margalha.miranda@gmail.com
Received for publication: 10.10.2018. Accepted in revised form: 27.09.2019

