











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
The C3 glomerulopathies (C3G) are a rare group of kidney diseases with an estimated incidence of one to three cases per million per year.1 The term C3G was adopted by expert consensus in 2013 with the recognition of two subgroups, dense deposit disease (DDD), and C3 glomerulonephritis (C3GN).2
Its pathogenesis involves an excessive activation of the alternative complement pathway, resulting in C3 fragmentation and deposition in glomeruli.1,3,4 The deposition of complement factors drives glomerular inflammation, resulting in a proliferative glomerulonephritis.3
The genetic, acquired or immunologic defects in regulators of alternative complement pathway, such as complement factor B (CFH) and I (CFI) and membrane cofactor protein (MCP, also called CD46), could lead to the hyperactivity of this pathway.5,6
The C3G presentation ranges from hematuria, proteinuria (with or without nephrotic syndrome), hypertension, nephritic syndrome to rapidly progressive glomerulonephritis. Serum C3 levels are low in most patients.1,6 Furthermore, C3G is often associated to an infection, usually an upper respiratory tract infection. The defect in the regulating mechanisms of alternative complement pathway prevents its downregulation following resolution of the infection, so the consequence is the excessive deposition of complement proteins and their breakdown products in the glomeruli.6 Careful review of the history can reveal exacerbation of renal disease during episodes of reinfection.3
The differential diagnosis with post-infectious glomerulonephritis (PIGN) can be a real challenge since it may have a very similar clinical and pathological features. Nonetheless the diagnostic is determined by patient’s clinical course and laboratory findings. PIGN is an immune complex-mediated glomerulonephritis triggered by an infection. Patients with PIGN usually present urinary abnormalities and C3 consumption that resolve within a few weeks without specific therapeutic intervention.5
C3G is also associated with the development of retinal drusen and atrophy, making an ophthalmologic evaluation an essential part of na C3G patient’s regular care.7 The underlying pathogenesis is poorly understood and clinical course does not appear to correlate with renal disease.6
Renal biopsy is required to establish the diagnosis of C3G. Immunofluorescence (IF) microscopy is essential to unequivocal demonstration of glomerular C3 staining of at least two orders of magnitude greater intensity than for any other immune reactant. Electron microscopy allows distinction between DDD and C3GN according to characteristics of deposits and specific localization pattern.4,6
Investigation of the complement system is indicated including assessment of complement activity, complement protein levels and screening for autoantibodies. Autoantibodies against complemente convertases or specific complement proteins are common, C3 nephritic factors (C3Nefs) are reported in 50%-80% of DDD patients and 44%-50% of C3GN patients.7 Genetic testing should be considered, since 25% of patients with C3G carry mutations in the genes encoding CFH, CHI, C3 or complement factor H-related proteins (CFHR1-5).5 Patients with more than 50 years should be evaluated for the presence of monoclonal gammopathy.1,7
The treatment of C3G is still a challenge and no disease-specific treatments are available. Treatment recommendations are based on case reports, series of cases and a few open-label trials.5,6
CASE REPORT
A 31-year-old African descent female presented to the emergency department with a history of asthenia and edema with one week of evolution. When asked about other symptoms, the patient also reported foamy urine and tonsillitis three weeks before the onset of symptoms.
The patient had prior history of recurrent lower urinary tract infections and an episode of acute pyelonephritis with acute kidney injury during pregnancy (serum creatinine maximal value 4.7 mg/dL with fully recovery of renal function in two weeks - sCr 0.7 mg/dL, without hematuria or proteinuria, at 23 weeks of gestation), three years before.
Pregnancy and delivery (at 39 weeks) took place without other complications and the child was born healthy. The patient had no other relevant personal history and there was no family history of kidney disease.
Physical examination revealed blood pressure of 136/90mmHg, periorbital edema and moderate peripheral pitting edema. The remainder of the physical examination was unremarkable.
On admission, laboratory testing revealed anemia (hemoglobina 10.4g/dL), acute kidney injury (sCr 2.0 mg/dL and urea 49 mg/dL), serum albumin of 2.3 g/dL and dyslipidemia (total cholesterol 254 mg/dL, LDL 181 mg/dL and triglycerides 130 mg/dL). The white blood cells and platelets counts were normal, C-reactive protein, sérum electrolytes, blood glucose, coagulation and liver-function tests were within normal range. Urinalysis revealed microhematuria [>20 red blood cells per high-power field(hpf)], leukocyturia (>20 leukocytes/hpf) and nephrotic range proteinuria (protein/creatinine ratio 9.7 g/g).
The etiological investigation of nephrotic syndrome showed that the autoimmune tests (anti-dsDNA antibody, ANA, ANCA, ENAs, rheumatoid factor and antiphospholipid antibodies) were negative. Serum level of C3 was decreased (27 mg/dL, normal range 90-180 mg/dL), but levels of C4 were normal. Serologic testing to human immunodeficiency virus, hepatitis B virus and hepatitis C virus were negative. Antistreptolysin O titer, immunoglobulins, serum free light chains, electrophoresis and immunofixation were normal.
Renal ultrasound showed normal-sized kidneys and slightly increased echogenicity of the renal parenchyma with preservation of corticomedullary differentiation.
The patient subsequently underwent a kidney biopsy which showed membranoproliferative glomerulonephritis pattern with crescentes (4/14 glomeruli - 3 cellular crescents and 1 fibrocellular crescent) on light microscopy, with mild chronicity changes (Figs. 1 and 2). Immunofluorescence microscopy revealed positive staining for C3 (++++) in the capillary wall and mesangium (Fig. 3). Meanwhile, IgA, IgG, IgM, C1q, K light chain, and L light chain staining were negative. Additionally, ultrastructural examination identified deposits primarily in the subendothelial and mesangial area, and rare subepithelial deposits (Figs. 4 e 5).
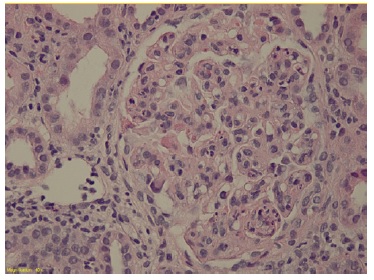
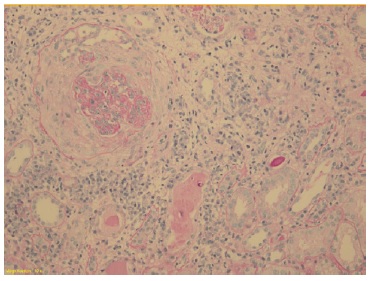
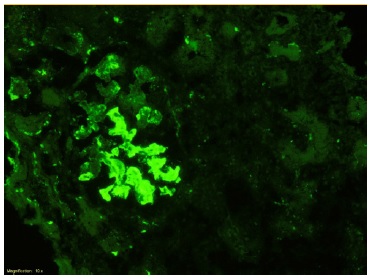
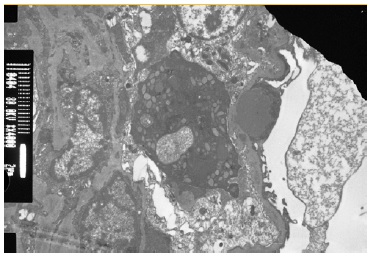
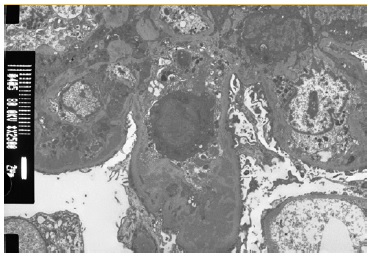
To address the nephrotic syndrome, she was treated with angiotensin-converting enzyme (ACE) inhibitor (lisinopril 5 mg/day), statin (atorvastatin 20 mg/day) and diuretic (furosemide 40 mg/day). Three weeks after the onset of symptoms, there was a favorable clinical and laboratorial response with normalization of the complement value and serum albumin and a significant reduction in proteinuria (protein/ creatinine ratio 1.1).
During follow up, there was maintenance of microscopic hematúria and proteinuria (lowest value P/C ratio 0.6 g/g) with stable kidney function corresponding to an estimated glomerular filtration rate of 78 mL/min/1.73 m2 (CKD-EPI). Considering these data, evaluation of the alternative pathway of complement was done. Genetic study did not reveal mutations in genes encoding C3, factor H (CFH), factor I (CFI), factor B (CFB) or complement factor H-related (CFHR) proteins. C3 nephritic factor (C3NeF), CFH, CFB and factor H autoantibodies were normal. Complement factor I deficiency (1.3 mg/dL, normal range 3.2-8.8 mg/dL) was diagnosed.
Based on the clinical history, histologic findings and laboratory tests, the diagnosis of C3GN associated with CFI deficiency was established. Nine months after symptom onset, the patient developed a new upper respiratory infection accompanied by nephrotic proteinuria (P/C ratio 4.9 g/g), macroscopic hematuria and hypoalbuminemia without worsening of kidney function. After treatment and resolution of the infection, glucocorticoid (prednisolone 20 mg/day) and immunosuppressive therapy (mycophenolate mofetil 2 g/day) were added to supportive care.
Pneumococcal conjugate vaccine (PCV13) and pneumococcal polysaccharide vaccine (PPSV23) were administered.
There was improvement of proteinuria (lowest value P/C ratio 0.4 g/g) and renal function remains stable (estimated glomerular filtration rate of 102 mL/min/1.73 m2). However, during follow-up, the patient still presented new episodes of mild infection with transient worsening of proteinuria (highest value of P/C ratio of 2.5 g/g), and no detected change in kidney function.
DISCUSSION
We report a case of a young women with C3GN associated with CFI deficiency. C3G may be diagnosed in both children and adults (with an average age of onset of 25 years), although mean age at diagnosis is lower for patients with DDD.6 The disease is heterogeneous and the spectrum of clinicopathological findings, triggering events, complement abnormalities and renal outcomes is wide.3
The most common presentation of C3GN is proteinuria and hematúria with relatively preserved kidney function.1 In our patient, the clinical presentation with nephrotic syndrome following an episode of upper respiratory tract infection suggested the diagnosis of PIGN.
PIGN usually resolves spontaneously in a few days and has a favorable prognosis. However, the persistence of active urine sediment and recurrence of nephrotic proteinuria during episodes of reinfection, made the diagnosis of C3G more likely in this case.
The complement system is essential to both innate and adaptive immunity and plays a major role in the defense against pathogens and in host homeostasis.5 Complement activity is controlled by numerous proteins known as regulators of complement activity, which are present in the fluid phase or are membrane bound.7
In order to investigate the cause of C3G, CFI deficiency was identified in our patient. Factor I is the specific fluid phase inactivator of C4b/C3b. Factor I and its cofactors (factor H, C4b-binding protein, complement receptor 1 or CD46) control the C3 and C5 convertases of the alternative and classic pathways of complement.8,9
Complement factor I deficiency is associated with recurrent pyogenic infections (Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis), autoimmune diseases, and kidney diseases.8,9
The first case to report the association between factor I deficiency and glomerulonephritis was published in 1999.9 In 2019, Boudhabhay et al reports a rare case of C3GN occurrence in a kidney transplant recipient with homozygous CFI pathogenic variant. The patient showed C3GN typical pathological findings in the kidney allograft two years after transplantation. The disease recurrence was not associated with infections or significant changes in renal function during the fourteenyear follow-up.10
C3G seems to have a chronic course, with progression to end-stage renal disease (ESRD) in up to 50% of the adult patients.5,11 This poor outcome reflects the lack of disease-specific therapy for C3G, whereby treatment is mainly focused on the reduction of proteinuria and suppression of renal inflammation.7 The evaluation of proteinuria over time is a marker for disease progression and can be used to predict kidney outcomes.
Support treatment with ACE inhibitors or angiotensin receptor blockers (ARBs) are first line therapy for proteinuria and blood pressure control. The reduction of proteinuria in 50% during the first 6 to 12 months is associated with lower risk of kidney failure.12
Vaccinations can provide an additional layer of protection to help prevent potentially severe infections in these patients. Therefore, the immunization for Neisseria meningitidis, Streptococcus pneumoniae and Haemophilus influenzae has been recommended. The eventual use of prophylactic antibiotics should be considered individually.13
Immunosuppressive therapy with mycophenolate mofetil (MMF) plus corticosteroids is associated with significantly higher clinical remission and a lower risk of kidney failure.3,11 Patients with genetic mutations were more likely to achieve partial remission, whereas complete remissions were more common in antibody-mediated forms.11 The mechanisms of action of this immunosuppressive regimen and the reasons why the response is heterogenous, are not completely understood.11 Treatment with corticosteroids plus MMF should be considered in patients with proteinuria >1 g/day and hematuria or declining kidney function despite supportive therapy for at least 6 months.14
In our case, the worsening of proteinuria during the follow-up period, despite supportive therapy, motivated the start of immunosuppression.
Although spontaneous partial remission was seen at the first presentation, treatment with corticosteroids and MMF was started to reduce the risk of relapse and progression of the kidney disease.
Spontaneous remissions in C3G are rare and the disease may remain stable for several years, despite persistence of proteinuria, until progression to ESRD.6 Immunosuppression, particularly corticosteroids and MMF, may reduce the risk of relapse, decrease the severity and frequency of symptoms, and potentially preserve kidney function in the long term when compared to support treatment.11 However, our patient still presented episodes of worsening proteinuria associated with recurrent episodes of minor infections. Despite this, the literature shows that treatment with corticosteroids plus MMF is well tolerated, and the incidence of infections was lower when compared to other immunosuppressive regimes.11 Relapses after discontinuation of therapy are common and long-term treatment regimens have shown better results.11
Data supporting the role of plasma therapy in C3G is lacking, a few case reports describe encouraging outcomes when a mutated protein has been identified.5
Given the relationship between alternative complement pathway dysregulation and C3G, complement targeting therapies, such as eculizumab, are being studied as new therapeutic options.2 Eculizumab is a humanized monoclonal antibody that binds C5 and prevents the formation of the membrane attack complex. Anti-complement therapy can be used on a case-by-case basis in patients with proteinuria and disease progression refractory to immunosuppressive treatment with MMF.14 Eculizumab seems to be more efficient in cases of crescentic rapidly progressive C3GN, than in non-rapidly progressive forms.15
Optimal duration of eculizumab therapy remains to be determined, but limited duration of treatment may be sufficient.15 Avacopan, na oral C5a receptor inhibitor, was evaluated in patients with C3G in a phase 2 clinical trial (ACCOLADE trial), study results suggested a beneficial effect in delaying C3G progression.16
Based on the pathology of disease, other complement-modulating agents should be given in consideration. The factor D inhibitor, danicopan, has demonstrated a favorable safety profile but resulted in incomplete and inadequately sustained inhibition of alternative pathway, leading to lack of efficacy.17 Alternative complement pathway inhibition with iptacopan, a selective inhibitor of factor B, was associated with a reduction in proteinuria and C3 deposit scores, in a phase 2 study.18 APPEAR-C3G is a phase 3 trial that aims to evaluate the efficacy and safety of iptacopan compared to placebo and standard of care in patients with native C3G.18 DISCOVERY was the first trial studying pegcetacoplan, C3 inhibitor, in C3G patients (native kidneys) and showed reduction of proteinuria with stable renal function.19
Further studies like VALIANT (enrolling) and NOBLE (enrollment complete) are warranted to investigate the therapeutic potential of pegcetacoplan in the treatment of C3G.18
Data on transplantation outcomes is scarce, but recurrence in allograft recipients is common (~50% in C3GN and 80%-100% in DDD) and histological recurrence can be documented immediately after transplantation.7,19 Low complement levels, high levels of circulating autoantibodies (C3 nephritic factor and factor H autoantibody), rapid progression to ESRD in the native kidneys and living-related kidney transplantation are factors associated with increased risk of recurrence.20 Treatment of recurrence is challenging in the absence of disease specific therapy. Allograft loss occurs in about half of the cases, but the disease may have a heterogeneous long-term renal outcome.5,7
Currently, there are several ongoing and planned clinical trials using new complement modulators that might transform the treatment of this rare disease. However, clinical trials to test new therapeutic agentes are challenging to perform and their results are influenced by the heterogeneity of disease and the scarcity of patients.
In this case, it is important to maintain vigilance of renal function and proteinuria. Given the safety and good tolerability of immunosuppression with corticosteroids plus MMF, this therapy should be considered as the standard of care in C3G, until newer complementtargeted drugs become available. However, spontaneous remissions can occur and the risk-benefit should be accessed and discussed with the patient when starting immunosuppressive therapy. Patients with mild disease (proteinuria <1.5 g/day, hematuria, and normal kidney function) can be initially managed with supportive measures only.

