











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Minimal change disease (MCD), previously known as lipoid nephrosis or minimal lesion is the most common cause of nephrotic syndrome in children but it is also seen in 10% to 20% of adults with nephrotic syndrome. MCD can be secondary to systemic diseases such as lymphomas, drugs, and hypersensitivity reactions. In adults, the disease is usually secondary while in children, it is usually primary or idiopathic.1
MCD is more common in Asia and has a male predominance (approximately 2:1) in young children, difference that disappears in adolescents and adults.1 MCD arises from a histopathologic lesion in the glomerulus featured by the absence of visible alterations in light microscopy and immunofluorescence, but with diffuse foot process effacement on electron microscopy.2,3
The pathogenesis of proteinuria in MCD has not been fully understood. It is proposed that abnormal cytokines from T-cells and/or abnormal T-cell regulation by B-cells are implicated in the pathogenesis of this disease.
Recently, it has been reported that podocytes play a key role.2,3 The clinical course in children is often benign with a high rate of remission after corticosteroid treatment. The response rate in adults is lower, with 5%-30% of MCD adult patients not responding to initial steroid therapy.3
ANCA-associated vasculitis (AAV) is the most frequent cause of rapidly progressive glomerulonephritis in adults.4 AAV occurs more commonly in Caucasians, has a male predominance and the typical age of disease onset is between the fifth and the seventh decade of life but it can be seen at any age. Clinical features may vary from non-specific constitutional symptoms to specific symptoms depending on the organs involved.4,5
Clinical observations showed that AAV is a two-hit process in which ANCAs together with proinflammatory stimuli, most likely of infectious origin, are required for the induction of full-blown disease.5
The pathologic hallmark of ANCA-associated glomerulonephritis is a necrotizing and/or crescentic glomerulonephritis without significant immune complex deposition detectable on immunofluorescence or electron microscopy.6
The main fluoroscopy staining patterns are diffuse granular cytoplasmic (C-ANCA) and perinuclear (P-ANCA). The C-ANCA pattern is almost exclusively associated with antibodies against PR3 but the P-ANCA pattern can be caused by many proteins, mainly myeloperoxidase (MPO), but also cathepsin G, elastase, β-glucuronidase, and others.7,8
Specifically, MPO and PR3 are strongly associated with primary systemic vasculitis, including granulomatosis with polyangiitis, microscopic polyangiitis and eosinophilic granulomatosis with polyangiitis. Autoantibodies to PR3 are useful as a diagnostic and disease activity indicator in granulomatosis with polyangiitis (GPA).9,10
We report a case with PR3-ANCA positivity but no histopathological evidence of vasculitis. The clinical course and the renal biopsy findings were compatible with MCD. The patient was approached as such and had a good clinical response.
CASE REPORT
Eighty-six-year-old woman, with no significant medical or pathological history, and normal renal function (baseline serum creatinine 0.86 mg/dL), presented with asthenia, anorexia, unquantified weight loss, bilateral knee swelling, and dyspnea with a month of evolution and progressive worsening. She did not take non-steroidal antiinflammatory drugs (NSAID), or other possible nephrotoxic drugs.
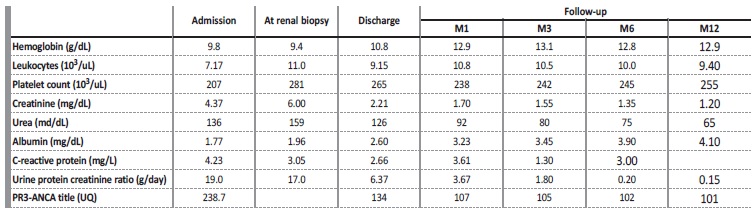
On admission, clinical examination revealed a malnourished patient (IMC 17 kg/m2). She was hypertensive (BP 180/100 mmHg), and reduced urine output (450 mL/24h). She had crackles on pulmonary auscultation, and generalized edemas. There were no obvious signs or symptoms of infection. Blood chemistry showed anemia de novo (Hemoglobin 9.8 g/dL; mean corpuscular volume 86.3 fl; mean corpuscular hemoglobin concentration 28.6 pg), severe renal impairment [serum urea (136 mg/dL), creatinine (Cr) (4.37 mg/dL)], and low serum albumin (1.77 mg/dL). Reactive C-protein and sedimentation rate were normal. Urinalysis showed proteinuria > 400 mg/dL with a protein-tocreatinine ratio of 19 g/g; microscopic examination of urinary sediment was normal.
Chest x-ray showed bilateral blunting of the costophrenic angle. There were no findings of cardiac enlargement or alveolar hemorrhage. Renal ultrasound was normal. Autoimmune screening using enzyme-linked immunosorbent assay (ELISA) revealed PR3-ANCA high titers (238.7 UQ). Serum complement C3 and C4 levels were in the normal range; anti-PLA2R and anti-glomerular basement membrane were negative. She had a decrease in IgG (620 mg/dL), normal IgA and IgM. Serum and urine electrophoresis were normal. Other autoimmune, inflammatory and infectious diseases were excluded.
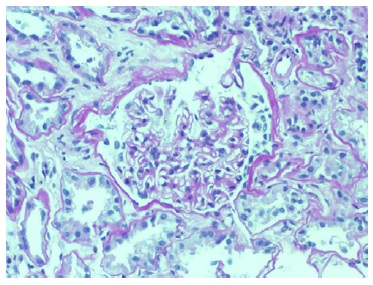
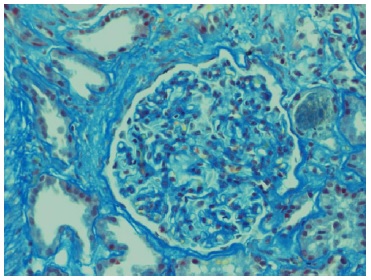
Echocardiogram had no signs of endocarditis. Screening for malignant diseases was negative: chest-abdomen-pelvis computed tomography scan showed moderate bilateral pleural effusion but absence of malignancy signs. Previous endoscopic studies were normal. The diagnosis of nephrotic syndrome was established. A renal biopsy was performed on day 12 of hospitalization. Until this period, there was no improvement in the renal function. Renal biopsy did not show any features of vasculitis or crescentic glomerulonephritis. The optic microscopy contained 47 glomeruli, 4 of which were globally sclerosed, and the remaining were optically normal.
Immunofluorescence (IF) was negative (Figs. 1 and 2). Unfortunately, electron microscopy was not performed.
The presumptive diagnosis of minimal change disease was made, and she started corticotherapy (methylprednisolone 500 mg/day IV, for three days followed by oral prednisolone 1 mg/kg/day), with clinical and analytical improvement (at one-month post treatment induction: sCr 1.70 mg/dL, protein-to-creatinine ratio 3.6 g/g; PR3 107) (Table 1). She had complete remission six months after therapy.
DISCUSSION
The measurement of ANCA antibodies plays an important role in the diagnosis of systemic vasculitis. However, the diagnosis relies on the presence of characteristic clinical findings.10-11
Recently, the presence of ANCA has been described in several other conditions such as inflammatory (ulcerative colitis, primary sclerosing cholangitis (PSC), primary biliary cirrhosis (PBC)), infectious (HIV, endocarditis), autoimmune diseases (autoimmune hepatitis (AIH), rheumatoid arthritis, systemic lupus erythematosus), or in association with some drugs.
Little is known about the pathogenesis of this diseases associated with ANCA, but it is believed that ANCA are capable of activate cytokine-primed neutrophils resulting in degranulation and endothelial cell damage.12-13
Knight et al, studied retrospectively a group of patients positive for cANCA/PR3-ANCA without vasculitis but with other diseases, and concluded that no patient developed vasculitis over a follow-up duration of 3-12 years (mean, 6.8 years), which may indicate that c-ANCA also reflects neutrophil-activating properties that are not specific to systemic vasculitis.9,12-13
In these cases, the antibodies can be false positives or directed against other proteins. Roozendaal et al, showed the presence of alphaenolase and catalase as target antigens for ANCA in inflammatory bowel disease. Orth et al, reported the presence of antibodies against ANCA in PSC, and Akisawa et al, reported the presence of autoantibodies directed against alpha-enolase in patients with PBC and AIH.14
As mentioned, false positivity for ANCA can be associated with non-vasculitic diseases.13-15 This “false positive” test is frequently associated with negative specific ELISA. Conversely, not all patients with systemic vasculitis are ANCA positive.16 Therefore, ANCA test results should be interpreted with caution.
Koderisch et al, suggested that false positive testing for ANCA and other autoantibodies, detected by indirect immunofluorescence techniques, may reflect non-specific IgG binding to Fc receptors, and in such cases, ELISA may prevent inaccurate diagnosis and treatment.8
McAddo et al, performed a retrospective analysis of 129 C and PR3-ANCA positive, and detected a higher-than-expected frequency (9.7 %) of “incidental” cases.17 Accumulating evidence supports that the induction of these autoantibodies seen in these cases could relate to the persistent antigenic stimulation of B-cells that leads to polyclonal immunoglobulin activation.
On the other hand, based on the observed homology between Staphylococcus aureus peptides and complementary PR3, it was suggested that the autoantibodies could result from molecular mimicry between infectious agents and ANCA antigens.18 Therefore, it is important to exclude infection in patients with symptoms and a clinical positive ANCA test.
More recently, Kain et al showed that infection with fimbriated gram-negative bacteria induces autoantibodies to human LAMP-2 through molecular mimicry that cause a new ANCA-subtype.19
Also, “incidental” PR3-positive may be due to increased use of ANCA testing in low clinical suspicion cases, and to the improvement of detection methods.17-19
A number of cases with an apparent overlap between true ANCApositive crescentic with other glomerular disease processes such as membranoproliferative glomerulonephritis, IgA nephropathy, membranous nephropathy, acute postinfectious glomerulonephritis, lupus nephritis, or diabetic glomerulosclerosis are being occasionally reported but the pathophysiology and the true meaning remains unknown.20
Recent data suggest that glomerular injury may be potentiated by synergistic effects of ANCAs and immune complex deposits. Jabur and Saeed, report a case with ANCA-positive necrotizing crescentic glomerulonephritis and nephrotic syndrome. The authors raise the possibility of whether the disease, at its outset, was a primary glomerulonephritis that later evolved to systemic vasculitis in the form of ANCA-positive necrotizing crescentic glomerulonephritis, or if they were two distinct disease entities.21
Our patient with MCD, presented also with a high title of PR3-ANCA even in the absence of vasculitis. The significance of PR3-ANCA positivity in the present case is unknown. We could not find previous reports of MCD associated with PR-3 positivity. We do not know if this is a false positive or if it reflects the existence of two independente pathological processes.
Our patient presented with rapidly progressive renal failure (RPRF), with nephrotic syndrome. The clinical presentation with RPRF associated with PR3- ANCA positive made the initial hypothesis of vasculitis more likely. However, histological findings and rapid response to corticosteroid therapy alone contradicted this hypothesis.
Also, clinical surveillance and future follow-up is necessary to monitor for signs of vasculitis development and PR3-ANCA levels tendency.
In conclusion, atypical clinical scenarios such as nephrotic syndrome with PR3-ANCA, can pose a diagnostic challenge. Renal biopsy is of value to establish an accurate diagnosis, delineate an appropriate treatment plan, and avoid inappropriately aggressive immunosuppressive therapy in patients without vasculitis. Long-term surveillance is essential.
Further studies are required to fully elucidate the pathogenic and clinical significance of PR3-ANCA positivity in MCD.

