











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Epidermal Growth Factor Receptor (EGFR) signalling is crucial for proliferation control and cellular differentiation, mainly of the epitelial cells, and is directly implicated in epithelial tumor development.1 It belongs to a tyrosine kinase family of receptors and binds to EGF and other factors on the cellular surface to induce cellular proliferation.2 Gain and loss-of-function studies of EGFR and its ligands have also demonstrated its role in magnesium and potassium renal regulation.3
Recently, an inherited homozygous mutation in EGFR with loss of function, has been reported in fatal cases of severe inflammatory skin disease and diarrhoea.1-3 We describe two further fatal cases of EGFR homozygous mutation in preterm newborns, both of consanguineous Roma parents, presenting electrolytic imbalances not previously reported. These cases, in continuity to those described in advance, clarify the difficult patient management and intends to reinforce the importance of genetic testing and, ultimately, finding new specific treatment to improve mortality outcomes.
CASE REPORTS
Case 1
At 25 weeks of gestation, a Romani teenager was referred to our Foetal Medicine Unit due to severe polyhydramnios and unilateral nephromegaly. A history of consanguinity was presente (first degree Romani cousins). Foetal ultrasound performed at 28 weeks revealed an increasing and tense polyhydramnios, megacystis and bilateral hydronephrosis, without obvious obstruction. After neuroprotection with magnesium sulphate and corticotherapy, an amnioreduction of 3600mL was performed. Bartter syndrome type I due to pathogenic variants in SCL12A1 gene was excluded.
An emergent caesarean section was performed at 30 weeks of gestation. The newborn scored 9 (1st minute) and 9 (5th minute) on Apgar and was admitted to the Neonatal Intensive Care Unit (NICU). Birth weight, length and head circumference were within 10th-50th percentiles (female Fenton growth chart).
On appearance, she looked immature, her skin was erythematous and friable, and presented global alopecia and hypotrichosis of the eyebrows (Figures 1 and 2 ). Over time, her skin progressively became thin and xerotic, with soft desquamation, and subcutaneous fat was absent (Figure 3).
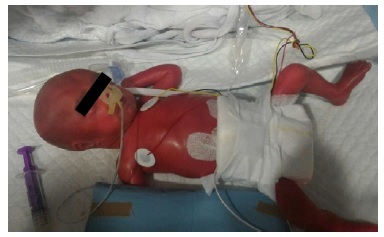
Figures 1 Skin appearance immediately after birth: erythematous and friable skin with global alopecia
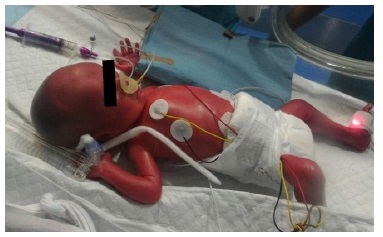
Figures 2 Skin appearance immediately after birth: erythematous and friable skin with global alopecia
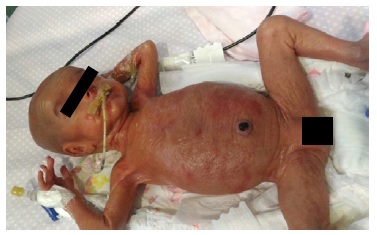
From the third week of age, she presented: intermittent polyuria (maximum 7.5 mL/Kg/h at 4th week of life); severe episodes of symptomatic hypernatremic dehydration (maximum 196 mmol/L, with seizures); hypokalaemia (minimum 2.6 mmol/L) with renal wasting of potassium (fractional excretion of K+, FEK+ 37%); severe hypomagnesaemia (minimum 0.85 mg/dL, with seizures) with hypermagnesuria (FEMg++ 44%); occasional hypercalciuria (urinary calcium/urinary creatinine, CaU/CrU=2 mg/mg); and normal chloride excretion. Maximum serum urea and creatinine were 49 mg/dL and 0.77 mg/dL, respectively. Bilateral nephromegaly was confirmed by ultrasound (40.4 mm and 41.9 mm) after birth. Subsequent ultrasounds showed both kidneys mildly enlarged and increased echogenicity with normal corticomedullar differentiation and no lithiasis.
General management with protective isolation, minimum manipulation with sterile sheets and gloves, intensive skin hydration with paraffin and barrier repair ointments and increased incubator humidity were implemented since birth. Daily chlorohexidine baths were also performed. Parenteral nutrition was required to maintain adequate fluid supply (210-260 mL/Kg/day), due to oral intolerance, supplemented on sodium, magnesium and potassium.
Nonetheless, cutaneous manifestations progressed to bleeding erosions, papules, pustules and abrasions predominantly in the face and neck. Skin biopsy was inconclusive, showing mild acanthosis, confluent parakeratosis and tissue infiltration with neutrophils and eosinophils. She presented recurrent sepsis, mainly methicillin-resistant Staphylococcus aureus (MRSA), Staphylococcus epidermidis and Extended Spectrum ß-Lactamase Producing Klebsiella pneumoniae requiring multiple cycles of broad-spectrum antibiotics.
Primary immunodeficiencies, such as IPEX-like (Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-linked), Omenn and Netherton syndromes, and syndromic ichthyosis were considered. However, immunoglobulin levels and lymphocyte count, subpopulations and proliferation assays were normal. Clinical exome and skin biopsy were performed.
Exome sequencing was performed, thereby identifying a likely pathogenic homozygous variant on EGFR gene, c1283G>A p.(Gly428Asp). Both parents are heterozygous for the same mutation. During the subsequent months she developed failure to thrive (birth weight achieved at the 37th day of life) and frequent vomiting, with normal stools. She began complementary food at five months of age with irregular tolerance. Due to parenteral nutrition dependency and persistent tubular dysfunction, she remained hospitalized until her death, which occurred at 11 months of age (cardiac arrest).
Case 2
The second patient was also a female born from consanguineous Roma parents (first degree cousins). Her 18-year-old mother was being followed in our Foetal Medicine Unit due to early severe intrauterine growth restriction (IUGR), <1st percentile, detected at 21 weeks of gestation. Foetal echocardiogram showed enlarged right cavities.
Amniocentesis followed by karyotype and CGH-array were performed, with normal results. Prenatal corticosteroid for lung maturation was administrated before emergent caesarian section at 33 weeks due to sustained foetal bradycardia. Apgar score was 9/9. Birth weight (895g), length and head circumference were below the 3rd percentile (female Fenton growth chart). Dysmorphic facial features were evident, with a triangular face, almond-shape eyes, short nose and inverted nostrils, and also severe skin alterations, such as generalised dermatosis with cutaneous fragility and erythema spots. As respiratory distress developed, she was admitted in the NICU and received CPAP until the 13th day of life.
She developed serious hydroelectrolytic imbalances from the first hours of life: severe polyuria (maximum 10 mL/kg/h at 11th day of life), leading to weight loss (23.5% of birth weight at 6th day of life); hypernatremic dehydration (maximum 167 mmol/L at 3rd day of life); hypokalaemia (minimum 2.2 mmol/L at 11th day of life) with renal wasting of potassium (fractional excretion of potassium, FEK = 35%); hypomagnesaemia (minimum 1.06 mg/dL at 19th day of life) with hypermagnesuria (FEMg 23.2%); hypercalciuria (CaU/CrU 3.8 mg/mg); and hypophosphataemia (2.9 mg/dL at 12th day of life). Maximum serum urea and creatinine were 62 mg/dL and 0.49 mg/dL, respectively.
High fluid rates (200-270 mL/kg/day) and parenteral nutrition with potassium, magnesium, calcium and phosphate supplies were prescribed to correct the hydroelecytrolytic imbalances, in addition to potassium and magnesium oral supplementation.
Neonatal echocardiogram confirmed enlarged right cavities that later normalized, with no need for cardiac care. Renal ultrasound was not performed and transfontanelar ultrasound showed no alterations. Primary immunodeficiencies and congenital ichthyosis associated with kidney disease were considered. However, immunoglobulin levels and lymphocyte count, subpopulations and proliferation assays were normal. Skin biopsy was postponed due to clinical instability related to multiple cutaneous sepsis. A likely pathogenic homozygous variant was identified in EGFR gene, c1283G>A p.(Gly428Asp), by exome sequencing. Parents were not studied.
General management was similar to described for case 1. In the following month, she developed failure to thrive (birth weight achieved at the 34th day of life) and frequent abdominal distension with irregular oral tolerance.
Recurrent sepsis with isolation of several microorganisms in blood cultures, conjunctival exudate and peri-catheter cutaneous exudate, mainly MRSA, Staphylococcus capitis, Enterococcus faecalis and Klebsiella pneumonia carbapenemase, required multiple and prolonged courses of broad-spectrum antibiotics. On the 70th day of life, she suddenly presented with bradycardia and hypoxaemia, refractory to advanced life support. Last blood culture, collected before death, was positive for Klebsiella pneumonia.
DISCUSSION
The differential diagnosis and therapeutic requirements of the described cases may be challenging. In case 1, the association of hydroelectrolytic imbalances (hypomagnesaemia, hypokalaemia, hypernatremic dehydration), nephromegaly and polyhydramnios raised the suspicion of tubulopathy. Bartter syndrome type I, caused by mutations in the SLC12A1 gene encoding Na-K-2Cl (-) cotransporter (NKCC2), was excluded by genetic amniocentesis. Bartter syndrome type IV, caused by mutations in the BSND gene, which encodes barttin, a subunit of the ClC-Kb and ClC-Ka Cl− channels, frequently presents with hydramnios at early stage and usually has a severe and persistente polyuria with increased chloride excretion and initial lack of hypercalciuria, as well as sensorial deafness, which were not observed in case 1. Another genetic condition responsible for hypomagnesaemia in the newborn/first infancy onset setting is EAST syndrome (Epilepsy, Ataxia, Sensorineural deafness, and Tubulopathy), also known as SeSAME syndrome (Seizures, Sensorineural deafness, Ataxia, Mental disability, and Electrolyte imbalance - hypokalaemia, hypomagnesaemia and metabolic alkalosis). This rare recessive disorder is caused by mutations in the KCNJ10 gene, which encodes K+ channel Kir4.1. Except for hypokalemia and hypomagnesaemia, none of the other clinical or laboratorial features were present in patient 1.4 However, none of these hypotheses explained cutaneous manifestations. At last, recessive isolated hypomagnesaemia, resulting from a mutation in EFG pathway, which combines hypomagnesaemia and inflammatory skin features, as described in case 1, was considered and later confirmed by genetic analysis. Diagnostic sequence in case 2 was shortened by its similarities with the first case and exome sequencing, performed sooner, also confirmed the diagnosis.
A recessive mutation in EGF has been associated with hypomagnesaemia, tetany, seizures and intellectual disability.5 Additionally, EGFR-targeting antibodies used for the treatment of colorectal cancer were found to cause magnesium wasting.6 Finally, EGFR loss of function was also previously described as a cause of hypomagnesaemia and neonatal skin and intestinal inflammation.1,2,7 ERBB family member epidermal receptor tyrosine kinase (EGFR) is composed of 28 exons and 27 introns. EGFR codes for 11 transcripts and 8 of them are protein coding. EGFR is a transmembrane glycoprotein that is expressed in many organs involved in diverse roles including proliferation, survival, embryogenesis and differentiation during development and maintenance of cellular physiology. EGFR’s loss of function was first described by Campbell et al.(1) in 2014, reporting an homozygous mutation c.1283G>A in exon 1 of the EGFR gene, resulting in a Gly428Asp substitution, from a Roma boy who died during infancy from neonatal inflammatory skin and bowel disease. A similar phenotype was posteriorly described by Ganetzky et al.(7) in 2015, in two preterm siblings from a consanguineous Roma family. Both found a reduced quantity of EGFR and poor response to functional tests, consistent with either an EGFbinding domain defect or a cytoplasmic mislocalization of mutante EGFR. Later, Alruwaithi and Sherlock (2018)8, Mazurova et al (2020)9 and Lemos M (2021)2, reported several cases with a consistente similar phenotype with neonatal onset (polyhydramnios, premature birth, severe skin disorder, recurrent sepsis, severe hydroelectrolytic imbalance, heart defects) and fatal prognosis. This rare disorder probably has a common ancestry (Roma origin), since all, but one, were reported in Roma patients. Hayashi et al (2018)(10) described the first case report of a biallelic compound heterozygous mutation in EGFR. This premature male of non-consanguineous Japanese parents shared similarities with the previously homozygous EGFR mutation patients including severe skin disorder, recurrent sepsis, diarrhoea, failure to thrive, electrolyte imbalances and, eventually, premature death at 4 months of age. However, he suffered from hyponatraemia and hyperkalaemia, the opposite of our patients and other previously described cases (table 1).
Table 1 Comparison of phenotype and hydroelectrolytic imbalances between case 1 and 2
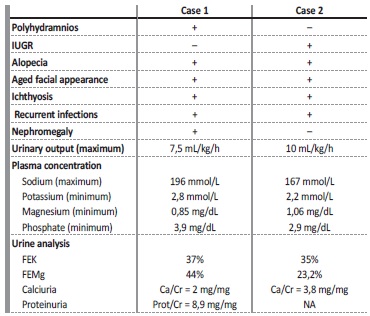
IUGR - intrauterine growth restriction; FE - fractional excretion; NA - not available
Renal involvement is not always present (only case 1 presented nephromegaly). Moreover, fluid intake and ion imbalances also vary in literature case reports. Our patients revealed partial oral tolerance, high fluid parenteral needs, hypokalaemia and hypernatraemia. Dehydration secondary to polyuria and transcutaneous loss caused severe hypernatraemia with seizures. During exacerbations, skin was more prone to infections, impaired cutaneous barrier function and increasing transcutaneous losses leading to hyponatraemia.
In particular, our cases had severe and difficult to manage hypomagnesaemia, without calcium disturbances. Electrolyte imbalances were reported in 3 patients1,2,10, specifically hypomagnesaemia in 2 of them1,2.
Magnesium is filtered freely at the glomerulus and reabsorbed in different percentages in the different segments of renal tubules.11 In the ascending limb of the loop of Henle (TAL) a common paracellular pathway, lined by CLDN16 and CLDN19, is responsible for concomitante reabsorption of Mg++ and Ca++, and loss of function in one of these proteins leads to hypermagnesuria and hypercalciuria. In distal convoluted tubule (DCT), magnesium reabsorption is transcellularly mediated, lined by TRPM6, an apical channel; loss of function of TRPM6 cause hypermagnesuria with normocalciuria.7 TRPM6 activity is increased after EGF stimulation.5,6. Therefore, in the absence of an effective EGF response, Mg++ reabsorption by TRPM6 channel is compromised and hypomagnesaemia develops.
In our patients, however, we found hypercalciuria. Several mechanisms may explain this unexpected finding. First, both patients were premature, and prematurity is associated with tubular immaturity and consequent hypercalciuria.14 Second, they both received parenteral calcium supplements, and a potential excess of calcium delivery might have been compensated through renal excretion. Third, hypomagnesaemia reduces parathyroid hormone (PTH) levels, thus preventing calcium reabsorption15; although we did not measure patients´ PTH, theoretically this can constitute another putative explanation for hypercalciuria. Fourth, a distal effect of urinary magnesium on renal calcium reabsorption was described, independent from PTH, in which TRPV5 can be inhibited by high urinary magnesium.16 Fifth, although TRPM6 conducts Mg++ at a much higher rate than Ca++ (5:1 ratio),17 its loss of function may contribute, at least in some extension, to calcium wasting. Sixth, hypophosphataemia, which was observed in both our patients, causes calcitriol synthesis, increasing intestinal calcium absorption and, consequently, hypercalciuria.18 And finally, gentamicin, which was prescribed several times for treatment of septic episodes, was recently found to block distal Ca2+ reabsorption by direct inhibition of the Ca2+ channel TRPV5.19 We believe that a combination of several of these mechanisms may possibly explain the hypercalciuria found in both patients.
Severe hypokalaemia observed, however, is also explained by EGFR mutation, through Mg++ TRPM6 channel dysfunction. Hypokalaemia associated with Mg++ deficiency is often refractory to treatment with potassium, despite the specific responsible mechanism not being completely understood20. Chou-Long Huang and Elizabeth Kuo reviewed the existing literature about this association and several
mechanisms were proposed. Na+/K+ ATPase function is compromised by hypomagnesaemia, with decreased cellular uptake of K+. When associated with increased urinary or gastrointestinal K+ excretion, as occurred in our patient, K+ wasting and hypokalaemia potentially occurs.20 Potassium is freely filtered at the glomerulus and its levels are precisely adjusted by balance between reabsorption in the proximal tubule and the loop of Henle and secretion in the distal convoluted tubule and the cortical collecting duct.20
Urinary K+ excretion occurs by two different apical K+ channels: Maxi-K channel, responsible for flow-stimulated secretion, and ROMK which is blocked by intracellular Mg++, limiting outward K+ flux.20 Even in healthy individuals, magnesium supplementation decreases urinary K+ excretion.20 EGFR blockage on enterocytes may disrupt the regulation pathway of chloride secretion, which would induce secretory diarrhoea.21 In contrast to other cases previously described, our patients showed no intestinal involvement.
Treatment can be very difficult. Although optimal management was initiated earlier in case 2, as we found multiple similarities to case 1 (family origin, clinical symptoms and laboratorial findings), fatal ending was observed. So far, the longest life expectancy described is 13 years of age.9 As in other reported cases in literature, our patients had premature death, consequence of multiple infectious complications
that ended in cardiac arrest before 12 months of age. Furthermore, our cases illustrate the persistent difficulties to manage these patients: recurrent life-threatening hydroelectrolytic disturbances, the need for high intakes of fluids, sepsis, severe cutaneous manifestations, and failure to thrive are challenging conditions, often resistant to treatment or requiring prolonged therapies.12,13
CONCLUSIONS
The authors aim to illustrate two rare cases of ichthyose-like rash and severe hydroelectrolytic imbalances with a vast differential diagnosis and its complex therapeutic management. EGFR mutations should be considered in infants with inflammatory, papulopustular or erosive skin disease with hypotrichosis of the scalp and eyebrows, diarrhoea and/or renal disturbances. Genetic analysis is crucial to confirm diagnosis and define prognosis. The links between EGFRdependent signalling and the regulation of skin inflammatory phenomena are still unclear. The total clarification of pathophysiology of this condition could help to identify a potential target for treatment of these patients.

