











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkCLINICAL PRESENTATION
We present the case of a 41-year-old South American female who was referred for nephrological evaluation due to persistent microscopic hematuria. Her past medical history included toxoplasma panuveitis and mild sensorineural hearing loss. There was no family history of chronic kidney disease (CKD). Laboratory investigation confirmed
microscopic hematuria with urinary sediment revealing dysmorphic erythrocytes. Renal function was normal (serum creatinine 0,62mg/dl) without proteinuria. Serologies for HIV and hepatitis B and C were negative, auto-immune investigation was negative and immunofixation excluded a monoclonal gammopathy. Renal and vesical ultrasound
were unremarkable and excluded possible causes for extra-renal hematuria. Despite benign evolution (patient maintained normal renal function and no urine abnormalities other than persistent microscopic hematuria) the patient expressed concern about the exact diagnosis of her kidney disease and a kidney biopsy was performed.
QUESTIONS
1. What is the most likely diagnosis considering the clinical presentation?
2. Considering the biopsy findings, what is the final diagnosis?
3. How should we manage the patient and what is her renal prognosis?
ANSWERS
1. What is the most likely diagnosis considering the clinical presentation?
In the presence of hematuria, it is important to evaluate nonglomerular causes such as infection, kidney stones, malignancy, gynecological or urologic origin1. In our patient the presence of dysmorphic erythrocytes, absence of other symptoms (weight loss, abdominal pain, dysuria) or risk factors for malignancy (smoking habits, exposure to carcinogenic agents) and exclusion of kidney stones or malignancy by ultrasound suggested glomerular origin of the hematuria.
Many diseases may present as asymptomatic urinary abnormalities and this is a common reason of referral for nephrological evaluation.
In this case the absence of proteinuria, hypertension or renal dysfunction indicated a probable benign prognosis. The most common causes of isolated microscopic hematuria are IgA nephropathy, Alport Syndrome and thin basement membrane nephropathy (TBMN). Other forms of glomerular disease may present with hematuria (infectionrelated glomerulonephritis, mesangioproliferative lupus, focal segmental glomerulosclerosis) but these are usually associated with other features/clinical progression distinct from the clinical presentation shown in this case.
IgA nephropathy is one of the most common glomerulopathies worldwide, and may present with a wide clinical spectrum, varying from isolated microscopic hematuria to synpharyngitic macroscopic hematuria, hematoproteinuria, renal impairment and progression to end stage kidney disease (ESKD). Diagnosis depends on kidney biopsy,
where deposition of mesangial IgA is the hallmark2.
On the other hand, Alport syndrome is a less frequent cause of hematuria and is associated with family history of kidney disease. In this disease there is a mutation in the collagen IV subunit leading to derangement of the glomerular basement membrane (GBM). Clinical manifestations include renal impairment evolving to ESKD, sensorineural deafness and ocular abnormalities2.
TBMN is also associated to a mutation in the collagen IV gene. It mainly manifests as an inherited disease and patients usually have normal renal function, minimal or no proteinuria and a family history of hematuria. Although patients usually have a benign disease course, a small percentage can evolve to ESKD. This entity was once considered a separate disease, but a recent position paper of the Alport Syndrome Classification Working Group proposed a new classification that considers both entities as a spectrum of the same disease (comprising mutations in the genes that affect the collagen IV molecule namely COL4A3, COL4A4 and COL4A5 genes)3. This new classification distinguishes 3 disease groups: 1) X-linked Alport Syndrome, which is caused by mutations in the COL4A5 gene whose male patients have a 100% risk of progression to ESKD; 2) Autosomal Alport Syndrome, which comprises patients with mutations in the COL4A3 or COL4A4 and includes patients with a high risk of progression to ESKD (namely those with recessive inheritance) and patients with milder forms (with dominant inheritance) including those classified as TBMN and 3) Digenic Alport Syndrome, where there is a mutation in 2 of the COL4A3, COL4A4 and COL4A5 genes with variable risk of progression to ESKD4 (Figure 1).
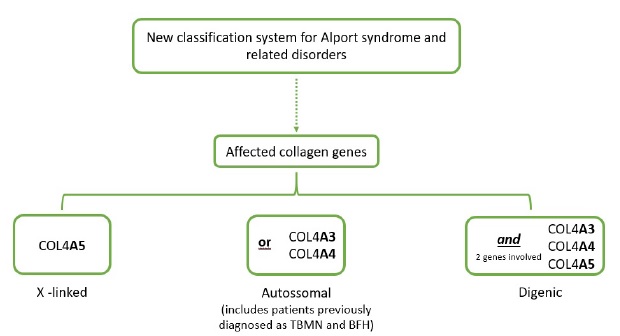
Adapted from Kashtan et al3. BFH, benign familial hematuria; TBMN, thin basement membrane nephropathy.
Differential diagnosis between these entities is not simple, although some clinical features may help. Hematuria in IgA nephropathy may be fluctuating and family history of isolated hematuria is infrequent. Alport syndrome, on the other hand, is usually associated with family history of kidney disease as well as extra-renal manifestations (as described above). In TBMN, a family history of hematúria is generally present and other renal/extra renal manifestations are infrequent.
In this patient, there was no history of familial CKD or ESKD (presence of familial hematuria could not be assessed) and the benign clinical evolution as well as absence of other renal manifestations (proteinuria or renal impairment) pointed towards a TBMN diagnosis.
Nonetheless, the presence of atypical features (mild sensorineural hearing loss) was a confounding feature that could be indicative of a disease in the Alport spectrum syndrome.
Although not performed in this patient, genetic testing as well as skin biopsy could aid in the differential diagnosis. The presence of a pathogenic COL4A5 variant confirms the diagnosis of X-linked Alport Syndrome. Genetic testing for autosomal recessive Alport syndrome is also useful and should be performed in patients whose clinical history suggests this disease. On the other hand, genetic testing specifically for diagnosis of TBMN (COL4A3 or COL4A4) is expensive and with few benefits. Exclusion of collagen mutation in the COL4A5 gene is more important in patients with asymptomatic hematuria because of the higher risk for progression to ESKD.
A skin biopsy could also be useful as the α5 chain is present in the epidermal basement membrane and its absence in the skin biopsy has high specificity for the diagnosis of X-linked Alport Syndrome.
Nonetheless a normal skin biopsy is not reassuring as, as stated above, patients with digenic Alport Syndrome may have a dismal renal prognosis and will have a normal skin biopsy.
2. Considering the biopsy findings, what is the final diagnosis?
The light microscopy sample was representative only of the medullary region. The immunofluorescence (IF) sample had four glomeruli and was negative (IgG, IgA, C3, kappa and lambda). Electron microscopy (EM) revealed one apparently normal glomerulus except for markedly reduced glomerular basement membrane (GBM) thickness, varying between 83nm e 167nm (reference values: 250-350nm) (Figures 2 and 3). Beyond GBM thinning there were no other changes (such as lamellation, irregular contours or heterogeneity of the GBM). The absence of light microscopy implies the loss of valuable information such as the exclusion of mesangial proliferation or segmental/global glomerulosclerosis (importante for IgA nephropathy). Nonetheless, negative IF suggests that this diagnosis is less probable. Also, EM did not show any mesangial deposits (with the limitation of a small sample since only 1 glomerulus was present). Considering biopsy findings and benign clinical evolution (patient with stable normal kidney function and no proteinuria for more than two years of follow-up) a diagnosis of thin membrane disease was made. It should be stressed that although X-linked Alport syndrome occurs with GBM lamellation, in early stages of the disease only thinning of the basement membrane may be present (although it usually occurs with intermitente GBM lamellation).
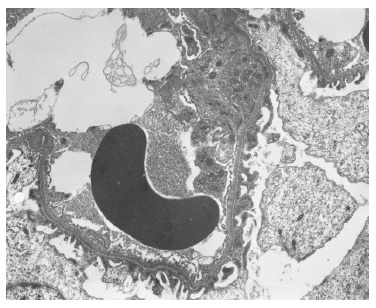
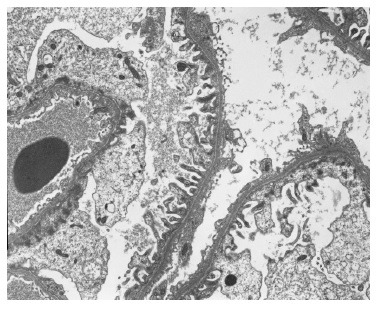
3. How should we manage the patient and what is her renal prognosis?
As stated above, the new proposed classification aims at promoting better treatment for patients, especially those previously thought to have benign disease. Patients with TBMN, now classified as Autosomal Alport Syndrome, usually have a benign disease course. Nonetheless, in a small percentage of patients, progression to ESKD is seen, as well as higher susceptibility in the presence of other kidney diseases.
In this case, since the patient did not have any risk factors associated with hematuria (namely proteinuria, renal impairment or hypertension), expert guidelines suggest annual monitoring of blood pressure, serum creatinine, urinary albumin-creatinine ratio and urinary protein-creatinine ratio 3.
Genetic test would be an important complement for the biopsy, confirming the diagnosis. This is important because the patient is still in reproductive age and since it is an autosomal dominant disease, the risk of disease in her descendants is high. Nonetheless, as stated above, genetic testing specifically for the diagnosis of TBMN is expensive and not usually performed, as testing for X-linked Alport syndrome is more useful because of its associated dismal kidney prognosis.

