










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.34 no.4 Lisboa dez. 2020
https://doi.org/10.32932/pjnh.2021.01.104
CASE REPORT
From light chain deposition to multiple myeloma – Case report and literature review
Ana Domingos1, Joana Vidinha1, Anabela Guedes1, Ana Macedo2,3, Mário Góis4, Pedro Leão Neves1,5
1 Nephrology Department – Centro Hospitalar e Universitário do Algarve – Faro, Portugal
2 Hematology Department – Centro Hospitalar e Universitário do Algarve – Faro, Portugal
3 University of Beira Interior – Faculty of Health Sciences – Covilhã, Portugal
4 Laboratory of Renal Morphology, Hospital de Curry Cabral, Centro Hospitalar e Universitário de Lisboa Central – Lisboa, Portugal
5 University of Algarve – Department of Biomedical Sciences and Medicine – Faro, Portugal
ABSTRACT
Monoclonal gammopathies consist of a broad spectrum of diseases, ranging from asymptomatic monoclonal gammopathy of undetermined significance to multiple myeloma (MM). Multiple myeloma is a malignant plasma cell disorder and accounts for 10% of all hematological malignancies and 1% of all malignancies.
Differential diagnosis may be challenging, considering the variety of clinical entities with similar behavior. About 15‑20% of MM only secretes monoclonal light chains, called light chain MM, which is associated with poorer outcome.
Two intermediate concepts were recently introduced, monoclonal gammopathy of renal significance (MGRS) and a wider concept of monoclonal gammopathy of clinical significance (MGCS). The former behaves as a clonal proliferative disorder with associated nephrotoxicity, but does not have the hematological criteria for MM, while MGCS expands this concept to other organs. A subtype of MGCS is monoclonal immunoglobulin deposition disease, a multisystemic disorder characterized by light or heavy chain deposition of monoclonal immunoglobulin in various organs and encompasses three clinical entities: Light‑Chain, Light‑and Heavy‑Chain, and Heavy‑Chain Deposition Disease (LCDD, LHCDD and HCDD, respectively).
We describe an unusual case of LCDD in which MM was subsequently considered although the proposed criteria are not met. We demonstrate the variability of clinical‑pathological presentation of LCDD, requiring a rapid decision‑making, particularly in terms of kidney and survival outcomes.
Key‑words: Monoclonal Immunoglobulin Deposition Disease, Light chain, multiple myeloma, kidney biopsy
INTRODUCTION
Monoclonal gammopathies consist of a broad spectrum of diseases, ranging from asymptomatic monoclonal gammopathy of undetermined significance (MGUS) to multiple myeloma (MM)1,2. Multiple myeloma is a malignant plasma cell disorder and accounts for 10% of all hematological malignancies and 1% of all malignancies3, with a worldwide incidence of approximately 7 cases per 100,000 persons per year.
Death rate of MM is 3.2 per 100,000 persons per year4, due to organ involvement. It is characterized by abnormal proliferation of plasma cells producing a monoclonal immunoglobulin (MIg), called monoclonal component (MC). They proliferate in bone marrow, resulting in severe skeletal destruction with osteolytic lesions, osteopenia and/or pathologic fractures, as well as anemia, hypercalcemia and susceptibility to infection5.
Signs and symptoms derive not only from bone invasion but also from renal toxicity, originated by the filtered paraprotein in renal parenchymal cells5. In general, the diagnosis is based on detection of monoclonal plasma cells, MC and related organ and tissue invasion6.
Up to 50% of MM patients present with kidney involvement7. Common patterns of renal involvement include cast nephropathy, light‑chain amyloidosis, monoclonal immunoglobulin deposition disease (MIDD) and acute tubular necrosis. Less frequent presentations consist of cryoglobulinemic, membranoproliferative or immunotactoid glomerulonephritis and fibrillary glomerulosclerosis or proximal tubulopathy.
Although rare, other presentations such as IgA, C3 or membranous nephropathy may also occur7,8. Two intermediate concepts were recently introduced: monoclonal gammopathy of renal significance (MGRS) in 2012 and a wider concept of monoclonal gammopathy of clinical significance (MGCS) in 20181,2,9,21. The former behaves as a clonal proliferative disorder with associated nephrotoxicity, but has no hematological criteria for MM, while MGCS expands this concept to other organs7. A subtype of MGCS is MIDD.
MIDD is a multisystemic disorder characterized by light or heavy‑chain deposition of MIg in various organs and encompasses three clinical entities: Light‑Chain, Light‑and Heavy‑Chain, and Heavy‑Chain Deposition Disease (LCDD, LHCDD and HCDD, respectively)11‑13.
The most common form of MIDD is LCDD (>70% of cases), with predominance of kappa (ĸ) light chains7. Multiple Myeloma is found in about 50% of LCDD or LHCDD cases and in 20% of HCDD cases, but these may occur in the absence of hematological disorder, which is defined as idiopathic [13‑15].
Although multisystemic, patients may be asymptomatic. Renal involvement usually dominates the clinical course, essentially with proteinuria (Pu) and chronic kidney disease (CKD)11,16.
Up to 50% of patients may have Pu in the nephrotic range, while about 25% present less than 1 g/day. Hematuria is also common. Usually there is a rapid decline in glomerular filtration rate (GFR), and MIDD may manifest as subacute tubulointerstitial nephritis or rapidly progressive glomerulonephritis (RPGN)13,17.
In another perspective, 15‑20% of MM only secrete monoclonal light chain, lacking expression of the normal immunoglobulin heavy chain. This subtype – Light Chain MM (LCMM) – is associated with poorer outcome18‑20.
We describe a peculiar case of ĸ LCDD in a patient with presumed MM, although classical criteria are not met10. We demonstrate the variability of clinical‑pathological presentation of LCDD, requiring a rapid decision making, particularly in terms of kidney and survival outcomes.
CASE REPORT
A patient, a 58‑year‑old white female, was first referred for a Nephrology consultation due to an increase in plasma creatinine (pCr), without previous history of CKD. Her personal background included low‑grade squamous intraepithelial lesion of the cervix (with high‑risk HPV detected) and repetitive tonsillitis and no other relevance to CKD.
Positive family history of breast and hepatic cancer is described, but no reference to CKD.
When questioned, the patient referred to foamy urine, nocturia and nicturia for 5 months and reported a 2/3‑month history of morning sickness and vomiting, sporadic diarrhea, weight loss of 8 kg and elevated blood pressure (160‑170/80‑90 mmHg). She also confirmed abundant water intake and only sporadic alcohol consumption; she is neither a smoker nor an illicit drug user. She had started taking omeprazole just few days before admission, with no other medication, and denied allergies.
The first pCr analysis from the general practitioner, 3 months ago, revealed a pCr of 2.04 mg/dL (from the basal value of approximately 0.8mg/dL) of unknown cause. Repeated control of one month showed a pCr of 2.17 mg/dL. This motivated a referral to the nephrologist and the mentioned consultation took place within a month.
Some previous analytical and imagological results were made available. Blood work showed hemoglobin of 10.5 g/dL (normocytic normochromic), erythrocyte sedimentation rate of 64 mm/h, uric acid of 7.9 mg/dL and a 2‑month urinary sediment with microscopic hematuria and Pu. Platelets, leukocytes, seric electrophoresis, complement, antistreptolysin O titer test (ASOT) and thyroid function were normal. Antinuclear antibody (ANA) and Anti‑double stranded DNA antibody (anti‑dsDNA) negative. An upper digestive endoscopy revealed H. pylori‑positive gastritis and a colonoscopy showed hemorrhoidal disease grade II/III. Abdominopelvic CT scan revealed a simple kidney cyst.
She was readily admitted to the hospital for further study. Relevant analytical results are presented in Table 1 and Figure 1.
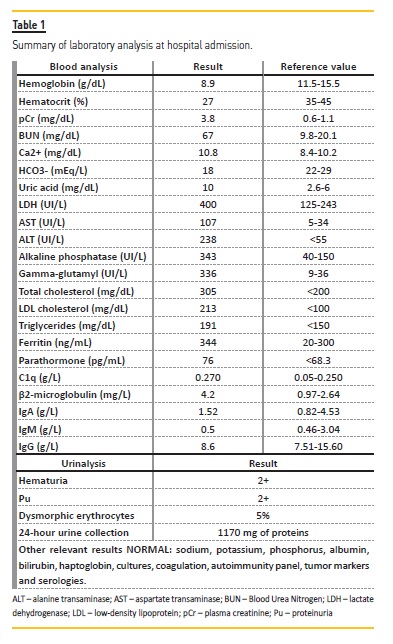
The patient soon presented with algic complaints on the left flank, nausea and vomiting. Extensive multidisciplinary approach was not conclusive. Ultimately it was assumed to be a difficult‑to‑manage muscular pain.
Her renal function deteriorated. Considering the hypothesis of acute tubulointerstitial nephritis, bearing in mind the out‑of‑proportion metabolic acidosis to renal dysfunction, a rapid progression and non‑specific symptoms, she was empirically administered prednisolone 1 mg/kg/day.
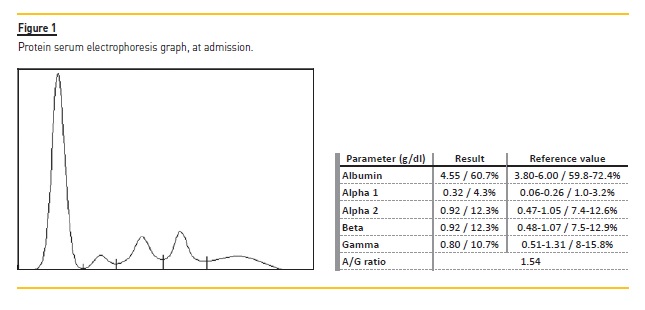
Kidney ultrasound revealed normal dimensions and maintained parenchymal‑sinus differentiation but slightly increased parenchymal echogenicity; an obstructive component was excluded. A kidney biopsy was performed. Among 9 glomeruli sampled for light microscopy, one was globally sclerotic. Glomeruli presented with mild increase in mesangial matrix, without cell proliferation. Tubular atrophy and interstitial fibrosis affected approximately 60% of the cortex. There was focal thickening of tubular basement membranes of nonatrophic tubules by PAS‑positive material (Figure 2A). Congo red negative. Immunofluorescence (IMF) revealed deposition of ĸ light chain in tubular basement membranes (Figure 2B), without lambda light chain (Figure 2C). It was only possible to perform serum free light chain (sFLC) determination later, showing a predominance of ĸ chains (794 mg/L) and a ĸ /λ ratio of 95.43 (N 0.26‑1.65).
Serum immunofixation showed a small MC in light ĸ chains (unmatched in heavy chains). Given the diagnosis of LCDD, bone marrow aspiration (BMA) was performed to evaluate an underlying plasma cell disorder. Myelogram was normocellular, with 4% plasmocytes and the fluorescent in situ hybridization (FISH) analysis for MM permitted the exclusion of genetic disorders.
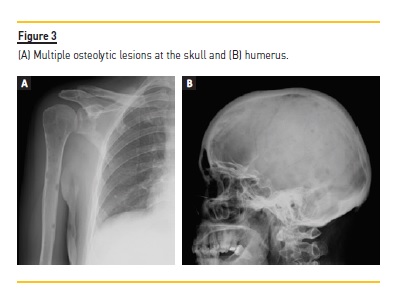
A skeletal X‑ray revealed multiple osteolytic lesions at the skull, humerus, shoulder blade and right femur (Figure 3). Magnetic resonance imaging highlighted the presence of more significant bone tumor infiltration at the level of C3, C5, C6, D6 and lumbar rachis.
A transthoracic echocardiography revealed left ventricle with septal hypertrophy, preserved systolic function but impaired relaxation and interatrial septal thickening, with no other relevant changes.
To evaluate potential respiratory involvement, in addition to the imaging study, respiratory function tests were performed, with normal results. Hematology collaboration was requested. The patient did not have clear criteria for the diagnosis of MM, but had target organ damage: anemia, hypercalcemia, and kidney and bone disease (CRAB)10. Thus, the diagnosis of ĸ LCMM was assumed (Durie‑Salmon stage IIIB, International Staging System stage II). Treatment with dexamethasone, bortezomib and thalidomide (VTD) was started3,4,12. The patient was discharged with follow‑up via external hematology and nephrology consultations. At discharge, she had pCr of 2.7 mg/dL and blood urea nitrogen (BUN) of 57 mg/dL.
She is currently under treatment and has been proposed for hematopoietic progenitor cell transplantation. Despite the patient not requiring dialysis during hospitalization and being under chemotherapy with good hematological results (including negative immunofixation after the second cycle of VTD), her renal function kept worsening (pCr 3.5 mg/dL, with an estimated GFR of 14 mL/min/1.73m2 by CKD‑EPI equation).
DISCUSSION AND CONCLUSIONS
This case reflects not only the challenge of a long differential diagnosis, but also the need for decision‑making as the research results emerged.
In a posterior analysis, the patient presented a rapid GFR decline, hematoproteinuria with only 5% dysmorphic erythrocytes, a subnephrotic Pu and a clinical course compatible with RPGN. These were, in fact, all consistent with the final diagnosis but still non‑specific.
Considering the vast differential diagnoses, the time required for multiple results to be ready and the lack of suggested clinical pain complaints (namely bone), this uncommon diagnosis became even more peculiar.
The presence of impaired kidney function with abnormal ĸ/λ ratio and plasmocytes <10% in the BMA raised the hypothesis of MGRS, since all criteria were met, according to the current definition (Table 2). This entity was first described in 2012 and redefined later in 2017 by the International Kidney and Monoclonal Gammopathy Research Group1. However, lesions presented by the patient were not restricted to the kidney. From another perspective, Fermand and colleagues2 introduced, for the first time in 2018, the concept of MGCS as an extension of the already known MGRS. This concept encompasses small B‑cell clones capable of producing severe manifestations through several mechanisms such as MIg deposition, cytokine‑mediated, auto‑antibody activity, among others2,9,21. Although the kidney is a frequent target, other organs may also be involved2,9. Unfortunately, unrecognized and underestimated, these small clones require aggressive therapy2. This concept fills the gaps of an asymptomatic MGUS and extents the concept of MGRS2. Monoclonal Immunoglobulin

Deposition Disease is now considered a subtype of MGCS, with deposition of MIg in a non‑organized fashion2.
The biggest controversy concerns the consideration of the definitive diagnosis of LCDD instead of LCMM. Studies show that in some cases routine electrophoretic techniques and immunofixation may not demonstrate a monoclonal protein in the serum (as most light chains are quickly filtered by the kidneys) or urine, although sFLC analysis will always detect it10,22. Because of the unavailability of our laboratory, some analyses were not performed, such as urine light chain screening and urine immunofixation, which could have given an early suspected diagnosis. However, we managed to get the correct diagnosis, with renal histology being essential. It is described that histological diagnosis of renal MIDD may precede the diagnosis of dysproteinemia in up to 70% of cases7,23.
Despite being an undesired diagnosis, ĸ LCD is associated with a better prognosis than λ. The high prevalence of MM in LCDD raised suspicion for this diagnosis. In this context we highlight that the patient did not have direct bone complaints, and this leaves open the question of her myalgia on the left flank, which, although apparently muscular, could have been a referred manifestation. Complementary study to date was negative. Only later and after presumed diagnosis was bone involvement demonstrated through programmed radiological study.
In addition, although most of these bone marrows present 10% or more clonal plasma cells, bone marrow involvement may not be homogeneous (a patchy disease) or aspiration may get diluted by peripheral blood; so there is a possibility, in 4% of patients, that bone marrow aspirate or biopsy show less than 10% plasma cells10,17. It may justify the results obtained as the patient meets the other criteria required.
Renal involvement by LCDD includes deposits in the tubular basement membranes and Bowman’s capsule (occasionally more prominent than those in the glomeruli)24. The location of the deposits defines the clinical presentation: renal insufficiency and mild Pu in those with tubular deposits and nephrotic syndrome when deposition is predominantly glomerular16. Inflammation and myeloma cast nephropathy may contribute to acute renal failure17,30.
Several authors describe liver involvement as frequent, with mild alterations in hepatic function, cholestatic pattern, possible life‑threatening hepatic insufficiency or portal hypertension13,17. The first two were present and complementary study came back negative (although liver biopsy was not performed). Soon after patient was admitted to hospital, her hepatic profile remained downward until normalization.
About a quarter of the patients will have cardiac manifestations; these may range from clinically asymptomatic to more severe manifestations, including cardiomegaly, dysrhythmias and severe heart failure13,17,26. Some reports suggest that LCDD‑induced cardiomyopathy may be reversible after successful treatment of the underlying plasma cells disorder25,26. Endomyocardial biopsy is the gold standard to demonstrate heart involvement in LCDD25.
Although rare, the respiratory tract may be involved. It is diagnosed by bronchial biopsy27, which was not performed in our case due to the absence of symptoms and the remaining tests being negative (including imaging and respiratory function tests).
Overall, the most probable diagnosis is ĸ LCDD in a patient with presumed MM / confirmed MGCS; CRAB criteria are present although lacking >10% of plasma cells in BMA.
Regarding treatment options, patients may or may not be eligible for autologous stem cell transplantation (ASCT), depending on risk stratification (Durie‑Salmon Staging and the International Staging System) and age. In those eligible, there is an induction therapy before stem cell harvest and, after that, patients can either undergo ASCT or resume induction therapy delaying ASCT until first relapse3. Regarding induction therapy, typically three to four cycles, several studies show that Bortezomib‑based triple therapy (namely VTD) has improved the survival of patients with mild to moderately reduced kidney function and those requiring dialysis3. Furthermore, achieving a complete response (CR) or very good partial hematologic response (VGPR) with chemotherapy has been shown to play an essential role in renal outcomes, delaying progression to ESRD and preventing recurrence of LCDD in those who receive a kidney transplant11. This idea was recently reinforced by Angel‑Korman and colleagues28 – although more data regarding outcomes of MIDD are needed, those who reach ESRD with a favorable response to treatments (CR or VGPR) should be considered for kidney transplantation.
In addition, renal response to treatment appears to have prognostic value for survival, with longer median survival among patients with renal recovery24. Autologous hematopoietic cell transplantation (AHCT) may be performed at all stages of kidney disease, including dialysis29.
Recent trials show that the median survival in MM is approximately 6 years. In those eligible for ASCT, 4‐year survival rates exceed 80%; the median overall survival is approximately 8 years3. There are few studies into median overall survival in LCMM patients (mainly in the era of the novel agents); however, LCMM seems to lead to a more aggressive disease and poorer outcome17,19. We believe that LCMM is an under‑diagnosed pathology, due to the possible manifestations and extensive differential diagnosis. Given its poor clinical course, severity and outcomes, clinical suspicion is essential.
References
1. Leung N, Bridoux F, Batuman V, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol. 2019;15(1):45–59.
2. Fermand J‑P, Bridoux F, Dispenzieri A, et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood. 2018;132(14):1478–1485.
3. Rajkumar SV. Multiple myeloma: 2018 update on diagnosis, risk‐stratification, and management. Am J Hematol 2018; 93(8):1091‑1101. [ Links ]
4. Gerecke C, Fuhrmann S, Strifler S, et al. The diagnosis and treatment of multiple myeloma. Dtsch Arztebl Int. 2016;113(27‑28): 470–476.
5. Kariyawasan CC, Hughes DA, Jayatillake MM, Mehta AB. Multiple myeloma: causes and consequences of delay in diagnosis. QJM. 2007;100(10):635. [ Links ]
6. Durie BG, Kyle RA, Belch A, et al. Myeloma management guidelines: a consensus report from the scientific advisors of the International Myeloma Foundation. Hematol J. 2003;4:379–398.
7. Korbet AM, Schwartz MM. Multiple Myeloma. JASN. 2006;17(9):2533–2545.
8. Leung N, Nasr SH. Myeloma‑related kidney disease. Adv Chronic Kidney Dis. 2014 Jan;21(1):36–47.
9. Bergstrom DJ, Kotb R, Louzada ML, Sutherland HJ, Venner CP. Canada M. Consensus Guidelines on the Diagnosis of Multiple Myeloma and Related Disorders: Recommendations of the Myeloma Canada Research Network Consensus Guideline Consortium. 2020;(July). [ Links ]
10. Caers J, Garderet L, Kortum KM, et al. European myeloma network recommendations on tools for the diagnosis and monitoring of multiple myeloma: what to use and when. Haematologica. 2018;103(11):1772–1784.
11. Sayed RH, Wechalekar AD, Gilbertson JA, et al. Natural history and outcome of light chain deposition disease. Blood. 2015;126(26):2805–2810.
12. Jimenez‑Zepeda VH, et al. Light chain deposition disease: novel biological insights and treatment advances. Int J Lab Hematol. 2012;34:47–355.
13. Ronco P, Aucouturier P, Moulin B. Comprehensive Clinical Nephrology 6th Edition – Renal Amyloidosis and Glomerular Diseases with Monoclonal Immunoglobulin Deposition. 6th Edition. Elsevier 2019;7:320–332.
14. Pant AD, Solez K. Light chain deposition disease in kidney: a review of the literature. J Pathol Nep. 2011;1:56–59.
15. Pozzi C, D’Amico M, Fogazzi GB. Light chain deposition disease with renal involvement: clinical characteristics and prognostic factors. Am J Kidney Dis. 2003;42:1154–1163.
16. Lin J, Markowitz GS, Valeri AM, et al. Renal monoclonal immunoglobulin deposition disease: The disease spectrum. J Am Soc Nephrol. 2001;12:1482–1492.
17. Salant D, Sanchorawala V, D’Agati V. Clinical conference: a case of atypical light chain deposition disease – diagnosis and treatment. CJASN 2007;2(4):858–867.
18. Rios‑Tamayo R, Rodríguez D, Chang‑Chan YL, Sánchez MJ. Epidemiology of Multiple Myeloma, Update on Multiple Myeloma. Available from: https://www.intechopen.com/books/update‑on‑multiple‑myeloma/epidemiology‑of‑multiple‑myeloma. [ Links ]
19. Rios‑Tamayo R, Sánchez MJ, García de Veas JL, et al. Light chain multiple myeloma: a single institution series. J Leuk. 2015;3:184. [ Links ]
20. Zhang JJ, Sun WJ, Huang ZX, et al. Light chain multiple myeloma, clinic features, responses to therapy and survival in a long‑term study. World J Surg Onc. 2014;12:234. [ Links ]
21. Merlini G, Stone MJ. Dangerous small B‑cell clones. Blood. 2006;108:2520–2530.
22. Perez‑Suarez G, Raya JM, Alvarez A, et al. Progressive renal failure as the first manifestation of monoclonal light‑chain deposition disease with rapid multiple organ involvement. Clin Nephrol. 2009;71(3):314. [ Links ]
23. Gallo GR, Lazowski P, Kumar A, Vidal R, Baldwin DS, Buxbaum JN. Renal and cardiac manifestations of B‑cell dyscrasias with nonamyloidotic monoclonal light chain and light and heavy chain deposit in diseases. Adv Nephrol. 1998;28:355–382.
24. Bladé J, Fernandez‑Llama P, Bosch F, et al. Renal failure in multiple myeloma: presenting features and predictors of outcome in 94 patients from a single institution. Arch Intern Med. 1998;158(17):1889. [ Links ]
25. Mohan M,Gokden M, Gokden N, Schinke C. A case of cardiac light chain deposition disease in a patient with solitary plasmacytoma. Am J Case Rep. 2016;17:173–176.
26. Garton MJ, Walton S, Ewen SW. Systemic lambda light‑chain deposition presenting with predominant cardiac involvement. Postgrad Med J. 1993;69:588–589.
27. Colombat M, Gounant V, Mal H, et al. Light chain deposition disease involving the airways: diagnosis by fibreoptic bronchoscopy. Eur Respir J. 2007;29(5):1057–1060.
28. Angel‑Korman A, Stern L, Angel Y, et al. The role of kidney transplantation in monoclonal ig deposition disease. Kidney Int Rep. 2020:5:485–493.
29. Badros A, Barlogie B, Siegel E, et al. Results of autologous stem cell transplant in multiple myeloma patients with renal failure. Br J Haematol. 2001;114(4):822. [ Links ]
30. Gu X, Herrera G. Light‑chain‑mediated acute tubular interstitial nephritis: a poorly recognized pattern of renal disease in patients with plasma cell dyscrasia. Arch Pathol Lab Med. 2006 Feb;130(2):165–169.
Ana Domingos, MD
Nephrology Department; Centro Hospitalar e Universitário do Algarve
E‑mail: atdomingos@chalgarve.min‑saude.pt
Disclosure of potential conflicts of interest:none declared
Received for publication: Aug 10, 2020
Accepted in revised form: Nov 16, 2020


{kind=link}