









 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
A doença granulomatosa crónica (DGC) é uma imunodeficiência primária caracterizada pela diminuição da produção de espécies reativas de oxigénio em células fagocitárias resultante de defeitos intrínsecos que afetam diversos componentes do complexo nicotinamida adenina dinucleótido fosfato (NADPH) oxidase. Os indivíduos afetados têm predisposição aumentada para infeções por bactérias e/ou fungos produtores de catalase e formação de granulomas em diferentes localizações 1.
Nos últimos anos assistimos a uma evolução relativamente à abordagem terapêutica do doente com DGC, para além da profilaxia antimicrobiana e do tratamento de complicações infeciosas e inflamatórias. Torna-se cada vez mais relevante, em todas as faixas etárias, avaliar individualmente a indicação/viabilidade de transplante de progenitores hematopoiéticos (TPH), uma opção terapêutica potencialmente curativa capaz de melhorar a qualidade de vida destes doentes 2.
Descrevemos neste trabalho a evolução clínica e a abordagem terapêutica realizada em quatro adultos com diagnóstico de DGC, em seguimento num Centro de Imunodeficiências Primárias (Tabela 1).
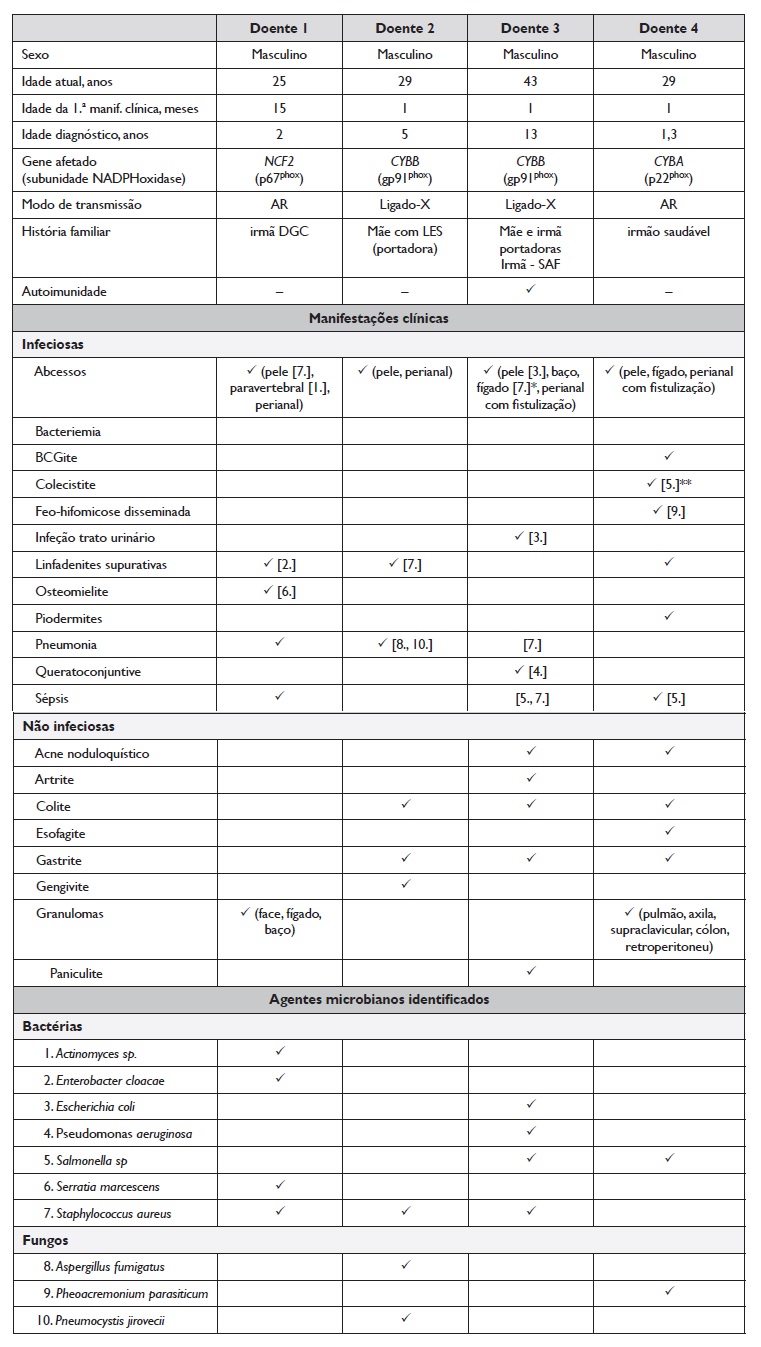
Os 4 homens, com idade atual compreendida entre os 25 e os 43 anos, apresentam defeito na capacidade oxidativa dos neutrófilos confirmado pelo teste do nitroazul de tetrazólio (NBT) e/ou teste da di-hidrorodamina (DHR). A análise genética permitiu a identificação de mutações no gene CYBB, que codifica a subunidade gp91phox da NADPH oxidase e responsável pela forma ligada ao X (DGC-X) em 2 doentes, e nos genes NCF2 ou CYBA que codificam as subunidades p67phox e p22phox, respetivamente, associados a formas autossómicas recessivas da doença (DGC-AR) nos restantes.
Em 3 doentes, as manifestações clínicas iniciaram-se no primeiro mês de vida, com adenite por BCG, sépsis a Staphylococcus aureus e doença inflamatória intestinal. O paciente 1 teve apresentação mais tardia, aos 15 meses, com linfadenite retroauricular por Enterobacter cloacae.
Embora com manifestações relativamente precoces, o diagnóstico ocorreu entre os 16 meses e os 13 anos, traduzindo-se num atraso de diagnóstico médio de 5,5 anos. Para este atraso deverá ter contribuído o facto de a DGC ser uma entidade muito menos reconhecida na comunidade médica à data de diagnóstico do doente 3, atualmente com 43 anos, mesmo apresentando manifestações clínicas muito típicas da doença.
Após o diagnóstico, todos os doentes iniciaram profilaxia com trimetropim-sulfametoxazol (TMP/SMX) e itraconazol, ainda que em 2 o início da profilaxia antifúngica tenha ocorrido mais tardiamente. Referir, também, que 2 doentes iniciaram, adicionalmente, profilaxia com Interferãoβ (IFN-β).
Apenas o doente 2, com DGC-X, foi submetido a TPH aos 9 anos de idade, a irmã a dadora. Quatro meses antes, o doente havia desenvolvido uma pneumonia grave por Aspergillus fumigatus e Pneumocystis jiroveci, com internamento em unidade de cuidados intensivos. Aproximadamente 12 meses após o procedimento houve perda do enxerto, justificando novo transplante, realizado menos de um ano depois, com dador de painel. Ambos os procedimentos foram realizados no centro onde o doente mantém seguimento desde o diagnóstico. Apesar do sucesso imediato, evoluiu posteriormente com doença de enxerto versus hospedeiro crónica cutânea, pulmonar e gastrointestinal.
Após 20 anos de seguimento, o doente mantém-se sem intercorrências infeciosas, mesmo sem profilaxias.
Os doentes 1 e 4 mantiveram-se sem intercorrências infeciosas graves durante a adolescência, mantendo os esquemas profiláticos. Nos últimos 5 anos, o doente 1 tem apresentado infeções graves, nomeadamente, abcesso
paravertebral por Actinomyces sp., abcesso do punho complicado com osteomielite por Serratia marcescens e, mais recentemente, pneumonia necrotizante por Aspergillus sp. complicada de fístula broncopleural e empiema de grande volume. O doente 4 teve nos últimos anos internamentos por descompensação de colite granulomatosa, tratada com corticoterapia oral e tópica (budesonido), e encontra-se desde há 18 meses sob terapêutica com voriconazol por feo-hifomicose disseminada por Pheoacremonium parasiticum.
O doente 3, atualmente com 43 anos, mantém-se sob tratamento profilático com TMP-SMX e itraconazol e sem infeções major, com exceção de episódio de queratoconjuntivite por Pseudomonas aeruginosa. Contrastando com esta estabilidade clínica, observou-se neste doente agravamento progressivo de linfopenia CD4 e perfil de imunossenescência precoce 3.
Após tipagem HLA e pesquisa de dador para TPH, apenas foi identificado dador de elevada compatibilidade (10/10) para o doente 4, estando em curso avaliação clínica para decisão da viabilidade deste procedimento a curto prazo. Não foi identificado dador compatível para os doentes 1 e 3.
O aumento da suspeição relativamente ao diagnóstico de DGC tem facilitado a identificação mais precoce destes doentes, que associada à implementação sistemática de profilaxia com TMP-SMX e itraconazol contribuiu para a diminuição da morbilidade observada nestes doentes e aumento da sua esperança média de vida 1,2. Estima-se que atualmente cerca de 90% dos doentes atinjam a idade adulta, mesmo sem recurso a TPH 1,2. As complicações infeciosas, particularmente fúngicas, e inflamatórias, permanecem como principais causas de mortalidade e morbilidade.
A estratégia terapêutica com intuito curativo, como o TPH, é hoje uma opção ponderada num número crescente de casos, mesmo em idade adulta 1,2. Embora com maior taxa de êxito em jovens, com menos complicações da doença (sobrevida a 3 anos 86,8% em crianças versus 76,4% em adultos) 2, as séries mais recentes de TPH em DGC reportam resultados cada vez melhores em adultos, mesmo naqueles com infeções recorrentes graves e complicações inflamatórias de difícil controlo 2. As recomendações internacionais para o TPH na DGC 4 indicam-no em doentes com infeções graves, evidência de disfunção de órgão progressiva (por exemplo, patologia restritiva pulmonar), ou ainda impossibilidade de acesso a cuidados de saúde diferenciados ou má adesão a terapêutica.
A terapia genética foi reportada internacionalmente por diferentes grupos num número muito limitado de doentes com DGC. Têm sido descritos avanços com diferentes modalidades de terapia genética, nomeadamente recorrendo a lentivírus ou a edição genética, sendo estas opções terapêuticas promissoras como alternativas ao TPH, com intenção curativa 1,2.
As complicações da DGC, principalmente a nível pulmonar e gastrointestinal, parecem ser mais graves no adulto 2. Estudos recentes revelam que os adultos com DGC mantidos sob terapêutica profilática/conservadora apresentam maior frequência de complicações infeciosas e não infeciosas do que indivíduos submetidos a TPH na infância na sequência de diagnóstico de DGC 5. A presença destas complicações condiciona piores resultados de TPH, ao contrário da idade do doente, que não influencia negativamente o êxito desta estratégia 5.
À luz do conhecimento atual, é reconhecida a mais-valia da realização de TPH tão precoce quanto possível, como estratégia potencialmente curativa em doentes com diagnóstico de DGC e disponibilidade de dador de elevada compatibilidade. Relativamente aos adultos em seguimento com DGC sob terapêutica conservadora, a presente reflexão pretende motivar a análise individual dos riscos e benefícios associados à realização de TPH, mesmo nos doentes com bom controlo clínico sob terapêutica profilática.